| ПОЗНАВАТЕЛЬНОЕ Сила воли ведет к действию, а позитивные действия формируют позитивное отношение Как определить диапазон голоса - ваш вокал Игровые автоматы с быстрым выводом Как самому избавиться от обидчивости Противоречивые взгляды на качества, присущие мужчинам Вкуснейший "Салат из свеклы с чесноком" Натюрморт и его изобразительные возможности Применение, как принимать мумие? Мумие для волос, лица, при переломах, при кровотечении и т.д. Как научиться брать на себя ответственность Зачем нужны границы в отношениях с детьми? Световозвращающие элементы на детской одежде Как победить свой возраст? Восемь уникальных способов, которые помогут достичь долголетия Классификация ожирения по ИМТ (ВОЗ) Глава 3. Завет мужчины с женщиной
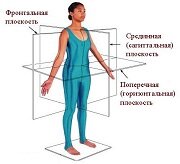
Оси и плоскости тела человека - Тело человека состоит из определенных топографических частей и участков, в которых расположены органы, мышцы, сосуды, нервы и т.д.
| Наследственные (моногенные) формы олигофрении
Метаболические олигофрении Фенилкетонурия Фенилкетонурия (фенилпировиноградная олигофрения, болезнь Феллинга) — наследственное заболевание обмена, характеризующееся преимущественным поражением ЦНС и прогрессирующим в первые 2 — 3 года жизни слабоумием. Фенилкетонурию впервые описал в 1934 г. A. Foiling. Позднее был установлен аутосомно-рецессивный тип наследования этого заболевания. В 1953 г. G. A. Jervis обнаружил, что в печени больных фенилкетонурией отсутствует фермент фенилаланингидроксилаза или резко снижена его активность. Н. Bickel, J. Gerrard и Е. М. Hickmans (1953) предложили лечить этих больных диетой с ограниченным содержанием фенилаланина. Частота заболевания мальчиков и девочек одинакова и составляет 1:10 000 новорожденных. Фенилкетонурия наследуется по аутосомно-рецессивному типу. Фенотипически здоровые родители больного ребенка являются носителями мутантного гена. Распространенность носителей гена фенилкетонурии в популяции составляет примерно 1:50. Для появления заболевания определенное значение имеет кровное родство родителей. Возникновение заболевания обусловлено наследственной неполноценностью гена, контролирующего синтез фермента фенилаланингидроксилазы, который обеспечивает реакцию превращения поступающего в организм с пищей фенилаланина в тирозин. В норме окисление фенилаланина в тирозин происходит в митохондриях печеночных клеток. Фенилаланин является незаменимой аминокислотой для построения белковой молекулы. Нарушение процессов превращения фенилаланина в тирозин приводит к резкому повышению содержания фенилаланина в сыворотке крови и спинномозговой жидкости (в норме содержание фенилаланина в сыворотке крови 1 — 2 мг%). Частично фенилаланин подвергается дезаминированию и превращению в фенилпировиноградную, фе-нилмолочную и фенилуксусную кислоты, которые наряду с фе-нилаланином в повышенной концентрации выводятся с мочой и могут быть легко обнаружены реакцией с 10%-ным раствором полуторахлористого железа. Дети, больные фенилкетонурией, рождаются с нормально сформированным и функционально полноценным головным мозгом, так как биохимические процессы плода осуществляются за счет обмена, происходящего в организме матери. Биохимические нарушения начинают развиваться сразу после рождения. Повышение уровня фенилаланина и его дериватов в сыворотке крови сопровождается снижением уровня других незаменимых аминокислот, а также вторичным нарушением углеводного, жирового и других видов обмена. Чувствительность нервной ткани к токсическому влиянию продуктов обмена фенилаланина, к дефициту гормонов и медиаторов нервной системы, а также к другим нарушениям обмена наиболее высока в раннем возрасте в связи с незрелостью ЦНС и повышенной проницаемостью гемато-энцефалического барьера. Патоморфологические изменения при фенилкетонурии проявляются в нарушении процесса миелинизации, глиозе и уменьшении массы мозга. Признаки заболевания обнаруживаются на первом году жизни. У одних детей уже в младенческом периоде отмечаются вялость, сонливость, слабая реакция на окружающее или беспокойство, у других — в первые месяцы развитие происходит нормально и только в 4—6 месяцев постепенно снижается реакция на окружающее, появляются повышенная раздражительность, пугливость, плаксивость. Дети становятся вялыми, задерживаются в развитии. Нередко у одних и тех же детей вялость и адинамия сменяются беспокойством и резким возбуждением. Довольно частым и ранним симптомом является рвота. Нередко первые признаки заболевания совпадают с введением прикорма и интеркуррентными заболеваниями, что иногда дает основание при ретроспективной оценке состояния ошибочно расценивать его как следствие перенесенных «стертых» менингитов и менингоэнцефалитов. Почти у всех детей наблюдается задержка физического развития: они отстают в росте, череп с признаками умеренной микроцефалии. Нередко отмечаются диспластические признаки (высокое нёбо, эпикант, деформация ушных раковин и др.). Около 80 —90 % больных — блондины со светлой, лишенной пигмента кожей и голубыми глазами. Примерно у 1/3 детей выявляются дерматиты и экзема, возникновение которых нередко совпадает с прикормом и неправильно расценивается как проявление экссудативного диатеза. У большинства детей отмечается повышенная потливость с характерным неприятным запахом пота. В неврологическом статусе чаще обнаруживается мышечная гипертония, повышение сухожильных рефлексов, гиперкинезы, тремор пальцев, рук, атаксия. Наблюдается также недостаточность моторики, координации и дифференциации тонких движений. Психопатологические нарушения при фенилкетонурии сложны и полиморфны. Степень интеллектуального дефекта колеблется от слабой выраженности до глубокой степени умственной отсталости. Имбецильность и идиотия отмечаются примерно у 90 % и дебильность у 10 % нелеченных больных. Особенностью психического недоразвития является фаза прогредиентной динамики в первые 2 — 3 года жизни. Затем процесс стабилизируется и постепенно обнаруживаются признаки эволютивной динамики. Структура интеллектуального дефекта имеет ряд особенностей, выявляемых в относительно редких случаях менее глубокой умственной отсталости: мышление инертно и недостаточно целе-направлено, отмечаются недостаточность сосредоточения и переключения внимания, плохая способность к активному запоминанию, большее недоразвитие гностических функций, основанных на анализе и синтезе пространственных представлений, при нередко несколько более сохранной формальной способности к обобщениям. У большей части больных имеется глубокое недоразвитие речи и дефекты произношения, коррелирующие с глубиной интеллектуальной недостаточности. В значительной степени страдает моторная функция речи. Отмечается склонность к эхолалии, персеверациям. При фенилкетонурии характерны нарушения в эмоционально-волевой сфере. Дети эмоционально однообразны, маловыразительны, не стремятся к эмоциональному общению с родителями и сверстниками. Их деятельность отличается недостаточной целенаправленностью, слабостью побуждений и интересов, однообразием при быстрой физической и психической истощаемости. Характерны нарушения контакта с близкими и сверстниками, безразличие к окружающему. У больных нередко отмечаются периоды психомоторного возбуждения, носящие психотический характер, — с импульсивностью, стереотипными вычурными движениями, манерностью, гримасами, эхопраксией и эхолалией. В структуре психического недоразвития большой удельный вес занимают также астенические и неврозоподобные нарушения: повышенная чувствительность и ранимость, истощаемость и утомляемость, расстройства настроения типа дистимий, страхов, заикания, энуреза и др. Диагностика фенилкетонурии проводится исходя из клинической картины и биохимических исследований. Предварительный диагноз устанавливается при помощи качественных проб на содержание в моче фенилпировиноградной кислоты, которая обычно появляется на 2 —3-м месяце жизни больного ребенка, в отдельных случаях — несколько позже. Для окончательной диагностики необходимо количественное определение содержания фенил-аланина в сыворотке крови. Гаргоилизм Гаргоилизм (синдром Гурлера) — группа заболеваний, обусловленных наследственной патологией соединительной ткани, различающихся характером обменных нарушений, но имеющих большое клиническое сходство. Характеризуются одновременным поражением ЦНС, органов зрения, опорно-двигательного аппарата, внутренних органов. Заболевание передается по аутосомно-рецессивному типу наследования или рецессивному. Мальчики заболевают в 2 раза чаще, чем девочки. Для возникновения синдрома имеет значение кровное родство родителей. В основе заболевания лежат генетически обусловленные нарушения обмена кислых мукополисахаридов, сложных белковополисахаридных образований, которые играют важную роль в структуре и функционировании соединительной ткани. У больных обнаруживается избыточное выделение их с мочой и накопление в соединительной ткани и внутренних органах. В связи с тем, что в различных органах и тканях преобладают разные типы мукополисахаридов, нарушения их обмена, особенно кислых гликозамин-гликонов, приводят к различным симптомкомплексам. Типичные клинические признаки заболевания проявляются в первые месяцы жизни ребенка. Характерен внешний вид больных: постепенно нарастают явления акромегалии, становятся грубыми черты лица, череп увеличивается в размерах, отмечаются дисплазии — лоб нависает над лицом, нос широкий с запавшей переносицей, деформированные ушные раковины расположены низко, губы сплюснуты, рост зубов неправильный, высокое нёбо, язык увеличен в размерах. По мере роста ребенка диспластические признаки становятся все более выраженными. Шея, туловище и конечности больных укорочены, грудная клетка деформирована, реберные дуги развернуты наружу, позвоночник искривлен; ладонь широкая, пальцы короткие, толстые. Отмечается малоподвижность суставов. При рентгенологическом исследовании выявляется уплотнение костей черепа, расхождение швов и истончение их краев, уплощение турецкого седла. Психическое развитие детей резко задерживается, характеризующие его процессы отличаются чрезвычайной вялостью и инертностью. Интеллектуальный дефект прогредиентно нарастает и чаще достигает степени идиотии. Выраженность клинических признаков, их частота и сочетания при мукополисахаридозах не всегда одинаковы. Диагностика гаргоилизма основывается на клинических, генео-логических и биохимических данных. Биохимическое обследование включает анализ мочи больных на кислые мукополисахариды. Отдельные фракции мукополисахаридов исследуются с помощью методов электрофореза на бумаге и хромотографии. |