| ПОЗНАВАТЕЛЬНОЕ Сила воли ведет к действию, а позитивные действия формируют позитивное отношение Как определить диапазон голоса - ваш вокал Игровые автоматы с быстрым выводом Как самому избавиться от обидчивости Противоречивые взгляды на качества, присущие мужчинам Вкуснейший "Салат из свеклы с чесноком" Натюрморт и его изобразительные возможности Применение, как принимать мумие? Мумие для волос, лица, при переломах, при кровотечении и т.д. Как научиться брать на себя ответственность Зачем нужны границы в отношениях с детьми? Световозвращающие элементы на детской одежде Как победить свой возраст? Восемь уникальных способов, которые помогут достичь долголетия Классификация ожирения по ИМТ (ВОЗ) Глава 3. Завет мужчины с женщиной
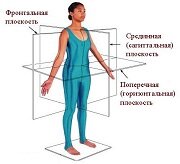
Оси и плоскости тела человека - Тело человека состоит из определенных топографических частей и участков, в которых расположены органы, мышцы, сосуды, нервы и т.д.
| ГИПЕРГИДРАТАЦИЯ НЕМЕСЕ СУЛАНУ. 12 страница
● кейбір зат алмасу өнімдері: асқын тотықтар, бос радикалдар -жатады. Соңғыларын аутомутагендер деп атайды. ТұҚЫМ ҚУАТЫН АУРУЛАРДЫҢ ЖІКТЕЛУЛЕРІ. Пайда болуындағы тектік ерекшеліктері мен қоршаған орта ықпалдарының арақатынастарына байланысты тұқым қуатын ауруларды үш үлкен топқа бөледі: ● нағыз тұқым қуатын аурулар. Олардың пайда болуында тұқым қуалаушылықтың маңызы өте зор, былайша айтқанда бұл аурулардың негізгі себепкер ықпалы болып өзгерген тектік ақпарат есептеледі. Ал, сыртқы орта ықпалдары бұл аурулардың пайда болуына әсер етпейді. Бұндай ауруларға бір геннің ауытқуларынан дамитын аурулар (фенилкетонурия, гемофилия, ахондроплазия т. б.) және хромосомалық аурулар жатады. ● екінші топқа гендік ақпараттың өзгеруінен дамитын тұқым қуатын ауруларжатады. Бірақ олардың клиникалық көріністері байқалуы үшін арнайы ортаның әсер етуі қажет. Мәселен, гетерозиготтық организмдерде орақ тәріздес жасушалы анемия көрінуі үшін дем алатын ауада оттегінің азаюы, тұқым қуатын гемолиздік анемия көрінуі үшін дәрі-дәрмектердің немесе басқа химиялық заттардың әсерлері болуы қажет. ● үшінші топқа көпфакторлық аурулар жатады. Оларға мысал ретінде, әсіресе ересек және ұлғайған адамдардың арасында кең тараған гипертензиялық ауру, жүректің ишемиялық ауруы, асқазан мен ұлтабардың ойық жарасы, қатерлі өспелер, туберкулез, пиелонефрит, қантты диабет т.с.с. ауруларды келтіруге болады. Бұлардың пайда болуында қоршаған ортаның қолайсыз ықпалдары негізгі себепкер ықпалдарға жатады. Олар тұқым қуалаушылыққа бейімділігі бар аурулар делінеді. Бұл аурулар тұқым қуатын ауруларға қарағанда өте жиі кездеседі. Олардың көріністері адамның жасына, жынысына, тамақтануына т. б. жағдайларға тығыз байланысты. Олар адам ауруларының 92℅-ын құрайды және олардың даму негізіндегі гендік өзгерістердің зерттелуі үлкен әдістемелік қиыншылықтарға тіреледі. Соған қарамай қазіргі медициналық тектанудың жетістіктері көптеген мүльтифакторлық аурулардың пайда болуындағы өзгерген гендердің орналасқан жерлерін табуға мүмкіншілік беріп отыр. Мәселен, асқазанның ойық жара ауруына әкелетін геннің АВО қан тобының жүйесімен байланыстылығы анықталды. Псориаз, гепатит, қантты диабет, тромбангаит т.с.с. аурулардың гені тіндік үйлесімділікті анықтайтын НLА — жүйесі антигендерімен байланысты. Сонымен бірге тұқым қуатын аурулар тектік (гендік) және хромосомалық аурулар болып бөлінеді. Тектік аурулар гендердің өзгерістерінен (мутациясынан) дамиды. Өзгерген гендердің санына қарай моногендік (бір гендік) және полигендік (көп гендік) аурулар болып ажыратылады. Моногендік аурулар бір геннің өзгеруінен дамиды және олар нағыз түқым қуатын ауруларға жатады. Полигендік аурулар тұқым қуалауға бейімділігі бар ауруларға жатады және көптеген ықпалдардың әсерлерінен дамиды. Хромосомалық аурулар хромосомалардың сандық және сапалық өзгерістерінен дамиды. Гендердің өзгерістері ұрпақтан ұрпаққа ауысатын болса, хромосомалардың өзгерістері ауыспайды. Өйткені соңғылары бар адамдар ұрпақ өрбіте алмайды. ТҮҚЫМ ҚУАТЫН АУРУЛАРДЫҢ ДАМУ ЖОЛДАРЫ. Түқым қуатын аурулар мына жағдайларда пайда болуы мүмкін: ● қалыпты тұқым қуатын ақпараттың болмауы; ● тұқым қуатын ақпараттың дерттік өзгерістерге ұшырауы; ● тектік құралдардың реттелулерінің бұзылыстары; ● бүлінген геномның дұрыс қалпына келмеуі. ДНК молекуласында мутагендердің әсерлерінен бір немесе бірнеше азоттық негіздер жоғалуы немесе олардың орны ауытқулары ықтимал. Сондықтан ДНК-дағы триплет жүйесінде тектік ақпарат және түзілген нәруыздың аминқышқылдық құрамы өзгереді. Осыдан қалыпты тұқым қуатын бағдарлама мүлде болмауы немесе дерттік өзгеріске ұшыраған болуы мүмкін. Тектік құралдардың белсенділігі оперонмен реттелінеді.Оперон оператор-геннен, реттеуші-геннен және бір немесе бірнеше құрылымдық гендерден тұрады. Реттеуші-ген оператор-генді реттейтін зат шығарады. Оператор-ген құрылымдық геннің белсенділігін арттырып немесе кемітіп тұрады. Сондықтан құрылымдық гендердің белсенділігі бұзылуы реттеуші гендердің мутациясынан болуьг мүмкін. Тіршілікте ДНК- молекуласы мутагендік әсерлерден мезгіл-мезгіл өзгеріп тұруы ықтимал. Бірақ, бүлінген молекулалардың қалыпты жағдайға қайта оралуына әкелетін бірқатар ферменттердің жүйесі бар. Бұл ферменттерге мыналар жатады: ● Фотореактивті фермент үльтракүлгін сәулелердің әсерінен болған ДНК-ның бүлінуін қалпына келтіреді. ● күңгірт реактивті жүйе: ♣ эндонуклеаза ДНК-молекуласының бүлінген жерін танып, оны үзіп алады. ♣ ДНК-полимераза ДНК-молекуласының үзіліп алынған бөлшек орнын жаңа бөлшектермен толтырады. ♣ полинуклеотидлигазалар фосфодиэфир байланыстарын құрастыру арқылы ДНК жіптерінің жаңа түзілген бөлшектерін қалпына келтіреді. Бұл ферменттердің түзілуін қадағалайтын гендердің бүлінуі тектік құрылымдардың көп жерлерінде тұрақсыздыққа әкеледі және жасушаларда ДНК- жіпшіктерінің бұзылыстарын өздігінен қалпына келтіру мүмкіншілігін жоғалтады. Адамда бүлінген ДНК-ны қалпына келтіретін жүйелердің гендік бұзылыстарының бірнеше түрлері белгілі. Мәселен, пигментті ксеродермия. Организмде эндонуклеазаның жоқтығынан (немесе жетіспеуінен) үльтракүлгін сәулесіне сезімталдық күшейеді. Өзгерген гендердің қалпына келуінің бұзылуы ихтиозда, атаксияда, пойкилодермияда, Даун ауруында, жүйелі қызыл жегіде т. б. дерттер кездерінде анықталған. Гендік аурулар. Гендердің мутациясы, хромосомаларға қарағанда, біршама аз мөлшерлерде болады және жиі әлсіздеу бұзылыстармен сипатталады. Аналық жасушалардың өсіп-өну қасиеті сақталады және сол себептен мұндай дерттер ұрпақтан ұрпаққа ауысады. Былайша айтқанда бұлар нағыз тұқым қуатын аурулар болып есептеледі, ал олардың тұқым қуалауы Менделдің заңына сәйкес келеді. Барлық гендік аурулар тұқым қуалау түрі бойынша аутосомдық-үстем (доминанттық), аутосомдық-бәсеңкі (рецессивтік), кодоминанттық, митохондриялық және жыныспен тіркескен түрлердеболады. Жыныспен тіркесіп тұқым қуалау ерекше тұрады және оларда Х- хромосомасымен тіркескен үстем, бәсеңкі (көпшілік жағдайда), Y-хромосомасымен тіркескен түрде берілуі мүмкін. Егер пайда болуына аллель жұбында бір дерттік басым ген жеткілікті болатын болса ондай ауруларды доминанттық тұқым қуатын аурулар дейді.. Ондай генді доминантты (үстем) ген деп атайды. Егер ауру аутосомды үстем генмен байланысты болса, онда аутосомды-үстем ауру дейді. Аутосомдық-үстем түрінде аутосомаларының біреуінде дертке ұшыраған ген қалыпты геннен басым болады да, гетерозиготтық организмде осы басым геннің белгілері барлық жағдайларда айқын көрінеді. Бұндай науқастардың шежіресін таратқанда ата-бабаларының үрім-бұтақтарының әрбіреуінде осындай аурулардың белгілері болғаны анықталады. Осы жолмен полидактилия, Альцгеймер ауруы, Гентингтон хореясы, ахондроплазия тоғышектің полипозы, отбасылық гиперхолестеринемия, нейрофиброматоз т.с.с. аурулар беріледі. Аутосомдық-үстем жолмен дерттің ұрпақтан ұрпаққа тарауына: ● еркектер мен әйелдерде дерттің бірдей жиілікпен кездесуі; ● ата-ананың әрбір буын ұрпақтарында дерттің болуы; ● жынысына қарамай туған балалардың екісінің бірінде дерттің кездесуі; ● сау туған балалары ары қарай дені сау ұрпақ өрбітуі – тән құбылыстар. Альцгеймер ауруыұрпақтан ұрпаққа тарайды. Бұл дертпен ауыратын адамдардың 40-жастан асқан соң ақыл-есі кеми береді. Олардың ми қыртысында, гиппокампында, ми бағанасында бүліністік өзгерістер болады. Альцгеймер ауруымен сырқаттанған адамдардың 21-жұп аутосомасында бета-амилоид түзетін нәруыз түзілуін қадағалайтын, немесе 14-жұп аутосомасында пресинилин-1 (PSN-1), немесе 1-жұп хромосомасында пресинилин-2 (PSN-2) түзілуін қадағалайтын өзгерген гендер болады. Осындай гендері бар адамдарда бета-амилоидтардың пептидтік тізбектері ұзарып, олар ми қыртысында т.б. ми құрылымдарында жиналып қалады. Оларды қарттық түйіндақтар немесе нейрофибрильдік шумақтар дейді. Осыдан нейрондардың бүліністері болып, олар тіршілігін жоғалтады, нейрондарда ацетилхолинтрансфераза ферменті түзілмеуінен ацетилхолин қатты азайып кетеді, түйіспелер арқылы жүйкелік серпіндердің тарауы бұзылады. Біртіндеп оттегі мен глюкозаның жүйке жасушаларымен пайдаланылуы азайып, мида қан айналым нашарлайды. Науқастардың жадында ұстау қабілеті тез арада немесе біртіндеп азая береді, жүре пайда болған шартты рефлекстер жоғалады. Гентингтон хореясы ауруымен еркектер ауырады. Олардың 4-жұп хромосомасында өзгерген ген болады. Осыдан мидың ГАМҚ-ергиялық нейрондары тіршілігін жоғалтып, дофаминергиялық нейрондары басым болып кетеді де, қаңқа бұлшықеттерінің дірілі және жан-дүниесінің бұзылыстары байқалады. Әйелдердің бұл аурумен ауырмайтыны олардың Х-хромосомаларының біріндегі ген 4-жұп хромосомадағы генді күшейтетін әсер етеді. Оны геномдық импритинг феномені дейді. Ахондроплазияаутосомдық-үстем жолменберілетін ауру. Ол аяқ-қолдың ұзындығына дамуы бұзылуымен, сүйектердің сынғыштығымен көрінеді.Дене ұзындығы және ақыл-есі дамуы әдеттегідей болады. Егер аурудың пайда болуына аллель жұбында екі өзгерген ген керек болса, ондай ауруларды бәсеңкі түрде берілетін аурулар дейді. Аутосомдық-бәсеңкіжолмен аурудың тарауы дерттік өзгеріске ұшыраған ген гомозиготтық организмдерде ғана көрінеді. Шежіре таратып талдағанда ұрпақтың барлық үрім-бұтақтарында дерт байқалмайды. Аутосомды-бәсеңкі берілетін аурулардың мынадай ерекшеліктері бар: ● еркектер мен әйелдердің арасында дерт бірдей жиілікпен кездеседі; ● еркектердің басқа әйелдерден туған немесе әйелдердің басқа еркектерден тапқан балаларында дерт кездеспейді; ● науқас адамның ата-анасының дені сау болады, ал оның басқа туыстарында дерт кездесуі мүмкін. Дені сау гетерозиготтық организмдер дерттік аллельдін иесі болады және екі дені сау гетерозиготты адамдардың некесінен ауру бала туады. Екі бірдей сирек кездесетін дерттік гендер бір аллельге өте сирек жиналады. Мұндай кездесудің мүмкіншілігі кездейсоқ некелердің (панмиксия) арасында он адамның біреуінде болуы мүмкін. Қан араласқан некелердің (инбридинг) арасында бұл мүмкіншілік 62,5 рет жиі байқалады. Бұл жолмен Фенилкетонурия, түссіздік, алкаптонурия, галактоземия, талассемия, гомоцистинурия, гипофиздік ергежейлік, микроцефалия, қояншық ауруының кейбір түрлері т.б. көптеген дерттер тарайды. Кодоминанттықжолмен ұрпақтан ұрпаққа берілгенде өзгерген және қалыпты гендер бірдей қызмет атқарады. Сондықтан орақ тәріздес жасушалы анемия кезінде әрі қалыпты гемоглобиндер, әрі орақ тәріздес гемоглобиндер болады. Дерттің көрінісі тек гипоксия кезінде байқалады. Бұл кезде S-гемоглобиндер тұнбаға ауысып, эритроциттердің гемолизін туындатады. Оттегінің қалыпты мөлшерінде гемолиз байқалмайды. Митохондриялық ұрпақтан ұрпаққа тарау митохондрийлардың ДНК молекуласында өзгерістердің болуымен байланысты болады. Содан олардың атқаратын қызметтері бұзылудан әртүрлі дерттер байқалады. Осы жолмен көру жүйкесінің атрофиясы, бұлшықеттердің тырысып селкілдеуімен көрінетін қояншық ауруы, митохондриялық миоэнцефалопатия, отбасылық дилятациялық кардиомиопатия дамиды. Бұл кезде дерт балаларына тек шешесінен ауысады. Сондықтан науқас әйелден туған балалардың барлығында дерт болады, ал науқас еркектен сау балалар туады. Жыныспен тіркесіпұрпаққа берілгенде өзгерген ген жыныстық, жиі, Х-немесе Y-хромосомада болады. Х-хромосомамен тіркесіп үстем түрде аурудың ұрпаққа тарауы Д-витаминіне төзімді мешелдік (рахит), Шарко-Мари-Тут ауруы кездерінде байқалады. Бұлай ұрпаққа тарауға: ●әрі еркектер, әрі әйелдер ауыруы, бірақ әйелдердің 2-есе жиі дертке ұшырауы; ●науқас еркектердің дерттік гендерін тек қыздарына ғана беруі; ●науқас әйелдердің дерттік гендерін әрі ұлдарына, әрі қыздарына таратуы; ●еркектердің әйелдерге қарағанда өте ауыр түрде сырқаттануы – тән. Х-хромосомасымен тіркесіп бәсеңкі түрде ұрпаққа тарауы түрлі-түстерді ажыратпаушылық (дальтонизм), сидеробластық анемия, гипогаммаглобулинемия, бүйректік глюкозурия, гемофилия А т.б. дерттер кездерінде байқалады. Бұл жағдайда: ● дені сау ата-анадан ауру бала туады; ● ер адамдар ауырады, олардың шешелері дерттік геннің тасымалдаушысы болады; ● дерттік ген әкесінен ұлға ешқашан берілмейді. Өйткені X хромосомы әкеден тек қыздарына ғана беріледі; ● дерттік гені бар әйелдерден туған ер балалардың 50%-нда дерт болуы мүмкін. Y-хромосомасымен тіркесіп ұрпаққа тарау азоспермия, саусақтардың ортаңғы буындарының сыртында және құлақ ішінде артық жүн өсу сияқты ауытқулар кездерінде байқалады. Бұл кезде олар әкесінен барлық ұлдарына ауысады. Гендердің қадағалауымен организмде әртүрлі қызметтер атқаратын нәруыздар түзіледі. Оларды: тіндердің, ферменттердің, гормондардың құрамына енетін нәруыздар - деп үш топқа бөлуге болады. Осыған байланысты гендік ауруларды үш түрге жіктейді: ● тіндердің құрылымына енетін нәруыздардың түзілуі бұзылуына әкелетін гендердің мутациясы жасушалар мен тіндердің құрылымын бұзады. Бұндай аурулар ұрпаққа әрқашан доминантты түрде беріледі. Оларға даму ақаулары: ахондроплазия, гемоглобинопатиялар (45-ке жуық түрі бар), эритроциттердің сфероцитозы, агаммаглобулинемия т. б. жатады; ● ферменттердің құрамына енетін нәруыздардың түзілуі бұзылуына әкелетін гендердің мутациясы. 1000-ға жуық ферментопатиялар белгілі. Осының нәтижесінде барлық зат алмасуларының бұзылыстары болады. Бұл аурулар әрқашан аутосомдық-бәсеңкі түрде беріледі және ауыр түрде өтеді; ● гормондардың құрамына енетін нәруыздардың түзілуі бұзылуына әкелетін гендердің мутациясынан гормон түзілуінің бұзылыстары болады. Бұл аурулар аутосомдық-үстем және бәсеңкі түрлерде беріледі. Мәселен, гипофиздік ергежейлілік аутосомдық-бәсеңкі, ал қантсыз диабет аутосомдық-үстем түрде беріледі. Сонымен бірге, геннің бүлінуі гормон түзілуінің кейбір сатыларына қатысатын ферменттердің жетіспеушілігіне әкелуі мүмкін. Мәселен: Гирке ауруы, фенилкетонурия, Вильсон-Коновалов ауруы; Ең жақсы зерттелген гендік аурулардың тобы — ферментопатиялар. Ферменттердің түзілуі бұзылыстарынан организмде: ● зат алмасуларының соңғы өнімдерінің тапшылыгы (альбинизм, гипотиреоз, гемофилия т. б.); ● зат алмасулары өнімдерінің ыдырамайтын заттарыңың жиналып қалуы (фенилкетонурия, гликогеноздар (Гирке ауруы), алкаптонурия т. б.) ; ● кері байланыс бойынша өзіндік реттелулердің бұзылыстарынан икемделістік серпілістердің тым артық болуы (қалқанша беадің гормондарының түзілуі бұзылғанда көрсетілген жағдайдың әсерінен зоб пайда болуы) т. с. с. құбылыстар байқалады.. ХРОМОСОМАЛЫҚ АУРУЛАР. Хромосомалық аурулар деп хромосомалардың құрамының немесе олардың санының өзгеруінен болатын ауруларды айгады. Олардың құрылымының өзгерістерін абберациялар (ауытқулар) дейді. Бұл өзгерістер жоғарыда көрсетілгендей ауру дамуына 4 жолмен әкеледі: ● қалыпты тұқым қуатын ақпараттың болмауынан; ● тұқым қуатын ақпараттың дерттік өзгеріске ұшырауынан; ● геномның реттелуінің бұзылыстарынан; ● бүлінген геномның дұрыс қалпына келмеуінен. Хромосомалардың абберациясының мәні хромосоманың кейбір бөлшектерінің сол хромосоманың ішінде немесе басқа хромосомалармен орын ауыстыруы болып есептеледі. Хромосомалардың мутациясының негізгі түрлері: ● делециялар — хромосоманың бір бөлшегінің жоғалуы; ● транслокациялар — әртүрлі хромосомалардың арасында үзінділермен (бөлшектермен) алмасуы; ● дупликация — хромосомалардағы кейбір гендердің екі еселенуі; ● инверсиялар — хромосома бөлшектерінің теріс (180°-қа) айналуы. Хромосомалардың құрылымдық өзгерістері негізінен көптеген ұрық даму ақауларына немесе ұрықтың өлуіне әкеледі. Қалыпты жағдайда жыныстық жасушаларда 23 жұп хромосома болады. Бір жұптың екі хромосомасы бір-бірінен ажырамағанда, қалыпты жағдайдан тыс, екі бірдей хромосома бір жасушада қалады да, оның саны 24, ал басқасында 22 хромосома болады. Егер ұрықтану кезінде гаметаның біреуінің бір хромосомасы кем болса, онда моносомды зигота пайда болады да, одан көпшілік жағдайда тіршілікке бейімсіз ұрық (эмбрион) дамиды. «У» жыныс хромосомасы болмағанда ғана өмірге бейімділік сақталады. 24 хромосомы бар гаметалар қалыпты 23 хромосомасы бар ұрык, жасушасымен ұрықтанғанда трисомияның себебі болады. Трисомиялы зиготалар 96% жағдайда өзінен-өзі түсіп қалады. Сол себептен барлық жаңа туған балалардың тек 0,3%-де три-сомия байқалады, ал оның ішінде 0,1%-де трисомия аутосомдық, 0,2% жыныстық хромосомалардың ажырамауымен байланысты дамиды. Аутосомдық трисомияның мысалына Даун ауруын (21-жұптың трисомиясын) келтіруге болады. Бұл ауру кезінде жүйкенің бұзылыстары, адамның ақыл-есі дамуының кемістігі, иммундық жүйенің ауытқулары байқалады. Науқас адамның хромосомасында өспе туындататын ген (онкоген) болуынан оларда жиі лейкоздардың дамуына қауіп-қатер болады. Бета-амилоид түзілуін қадағалайтын 21- жұп хромосоманың генімен жарыместік дамуын байланыстырады. 13-жұп хромосоманың трисомиясын Паттау синдромы деп атайды. Бұл кезде микроцефалия, қоянжырық, тесік таңдай, полидактилия, жақ сүйектерінің жетілмеуі, жүрек қақпақшаларының ақаулары байқалады. 18-жұп хромосоманың трисомиясын Эдвардс синдромы дейді. Бұл ауру құлақ қалқандарының пішіні өзгеруімен, көз саңылауының тарылуымен, астыңғы жақ сүйектің аз дамуымен, жүректің, бүйректің және ішек-қарын жолдарының ақауларымен көрінеді. Жыныс хромосомаларының трисомиялары үш «Х»-хромосомаларымен (Х-трисомия) немесе «ХХҮ-трисомия» (Клейнфелтер синдромы) түрлерінде байқалады. Бұл кездерде Х-трисомиямен ауыратын әйелдердің жасушаларында 2 жыныстық хроматин, Клейнфелтер синдромымен ауыратын ерлерде 1-жыныстық хроматин кездеседі. ХО-моносомия Шерешевский-Тернер синдромы делінеді және бұл әйелдерде жыныстық хроматин болмайды. ТҮҚЫМ ҚУАТЫН АУРУЛАРДЫ ЗЕРТТЕУ ӘДІСТЕРІ Бұл әдістердің ішінде ең кең тарағаны — шежіре тарату (генеалогиялық) әдісі. Осы әдіс бойынша науқас адамның ата-бабаларының және жақын туысқандарының арасында аурудың тарауы бірнеше буындарда қаралады. Егіздерді зерттеу өдісі де маңызды орын алады. Бұл әдіс аурудың пайда болуындағы тұқым қуалау мен сыртқы орта ықпалдарының маңызын салыстырып анықтауда үлкен үлес қосады. Егіздер әртүрлі болады. Бір ұрықтан таралған немесе монозиготты егіздер бір ғана ұрықтанған аналық жасушадан (бір зиготадан), екі эмбрион бөлініп дамиды. Олардың генотипі толық бірдей болады. Егер әйелдерде екі аналық жасуша бірден жетіліп екі аталық жасушалармен ұрықтанса, ондағы туған егіздер әртүрлі аналық жасушалардан дамиды. Оларды дизиготты егіздер дейді. Бұл егіздердің генотипі бөлек туған балалардағы сияқты әртүрлі болады. Егіздерді зерттеу әдісі бойынша бір және екі ұрықтан дамыған егіздердің өзара ұқсастығын салыстыру арқылы әрбір белгілердің дамуында тұқым қуалау мен қоршаған ортаның маңызын анықтауға болады. Бір белгімен біріне-бірі ұқсас егіздер жұбын конкордантты егіздер деп атайды. Егер егіздердің біреуінде бір белгі болып, екіншісінде болмаса, ондай жұпты дискордантты деп атайды. Бір ұрықтан дамыған егіздерде, екі үрықтан дамыған егіздермен салыстырғанда, ауру бірдей жиілікпен кездессе, ол аурудың пайда болуында тұқым қуалаушылықтың маңызы үлкен екендігін көрсетеді. Ал, олардың біреуі сырқаттанып, екіншісі сау болса, онда аурудың пайда болуы қоршаған ортаның ықпалдарынан дамуын дәлелдейді. Популяциялық әдіспен халықтардың кейбір топтарының арасында тұқым қуатын аурулардың тарауын, олардың зандылықгарын анықтауға болады. Цитологиялық әдіс арқылы бөліну сатыларындағы жасушалардың ядроларында кариотипті және жыныстық хроматинді микроскоппен тексереді. Биохимиялық әдіс бойынша гендік аурулардың биохимиялық ақауларын анықтауға болады. Бұл соңғы екі әдіс жатыр ішінде даму кезіндегі ауруларды анықтауда ерекше бағалы. Қағанақ сұйығын (амниоцентез) зерттегенде зат алмасуының әртүрлі өнімдерінің мөлшерін, ферменттердің қабілетін және жасушаның бөлшектерінде жыныстық хроматин мен кариотипті анықтайды. Осы жолмен 70-ке жуық тұқым қуатын ауруларды анықтауға және керекті жағдайларда нәрестені дер кезінде алып тастауға болады. Эксперименттік әдіс арқылы кейбір тұқым қуатын ауруларды зертханалық жануарларда алуға болады. Тұқым қуалау кемістігі бар жануарлардың таза буындары өсіріліп шығарылған. ТұҚЫМ ҚУАТЫН АУРУЛАРДАН АЛДЫН-АЛА САҚТАНДЫРУ ЖӘНЕ ЕМДЕУ ЖОЛДАРЫНЫҢ негіздері. Қазіргі жағдайда тұқым қуатын аурулардан сақтандырудың негізгі жолы болып ондай дерттері бар отбасыларын медициналық тектану емханаларда зерттеулерден өткізу есептеледі. Тек медициналық генетикалық білімі бар маман дәрігерлер ғана осы жолмен сапалы баға бере алады. Генетикалық емхананың негізгі мақсаты — ауру баланы тудырмау. Ол үшін гендік ауруы бар отбасында шежіре тарату, гендік жорамалдау тәсілі және керекті биохимиялық, цитогенетикалық ажыратулар арқылы ауру баланың туу мүмкіншілігін дәл есептеу керек. Тұқым қуатын ауруларды емдеу мүмкіншіліктері қазіргі уақытта берік орын алып келеді. Патогенездік емдеу әдістерінің ұстанымдары төмендегідей: |