| ПОЗНАВАТЕЛЬНОЕ Сила воли ведет к действию, а позитивные действия формируют позитивное отношение Как определить диапазон голоса - ваш вокал Игровые автоматы с быстрым выводом Как самому избавиться от обидчивости Противоречивые взгляды на качества, присущие мужчинам Вкуснейший "Салат из свеклы с чесноком" Натюрморт и его изобразительные возможности Применение, как принимать мумие? Мумие для волос, лица, при переломах, при кровотечении и т.д. Как научиться брать на себя ответственность Зачем нужны границы в отношениях с детьми? Световозвращающие элементы на детской одежде Как победить свой возраст? Восемь уникальных способов, которые помогут достичь долголетия Классификация ожирения по ИМТ (ВОЗ) Глава 3. Завет мужчины с женщиной
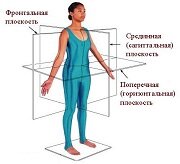
Оси и плоскости тела человека - Тело человека состоит из определенных топографических частей и участков, в которых расположены органы, мышцы, сосуды, нервы и т.д.
| Методы, применяемые для исследования состояния солевых систем.
Для изучения могут быть использованы различные методов зависимости от природы исследуемых систем. Одним из методов, применяемых при изучении конденсированных систем, является термический анализ, так как он сравнительно просто позволяет по изменению температуры (при отводе и подводе теплоты) получить общую картину взаимодействия веществ в системе. Для расшифровки природы фазовых превращений применяются и другие методы: рентгеноструктурный фазовый анализ, инфракрасная спектроскопия, магнитно-резонансные методы, микроскопические исследования, изучение электрических свойств и так далее. Из электрических свойств наиболее часто изучаются удельная электропроводность и ее температурный коэффициент. Большое значение имеют и такие методы как определение кристаллооптических свойств, изучение вязкости расплавов, исследование сжатия или расширения и некоторые другие. В настоящем пособии рассматриваются, главным образом, те методы, которые в дальнейшем будут применяться для построения диаграмм плавкости и растворимости систем. Физические и химические процессы, сопровождающиеся заметным выделением или поглощением теплоты, могут быть изучены путем измерения температуры при нагревании или охлаждении системы. Это является сущностью термического анализа. Существуют два основных вида термического анализа: 1. Термический анализ с визуальным наблюдением за температурой появления и исчезновения кристаллов, так называемый визуально- политермический метод. 2. Метод построения кривых время – температура: а) с визуальным отсчетом температуры; б) с автоматической регистрацией температур фазовых превращений с помощью саморегистрирующих приборов и построения кривых нагревания (охлаждения). Визуально-политермический метод Сущность этого метода заключается в наблюдении за появлением первых кристаллов, выделяющихся при охлаждении расплава, и исчезновением их при нагревании с одновременной регистрацией соответствующей температуры. Наблюдения за исчезновением и появлением кристаллической фазы при нагревании и охлаждении расплава производят до получения близких температурных данных, следующих непосредственно друг за другом. За температуру превращения принимают среднее арифметическое из двух последних измерений. Для обеспечения однозначности результатов охлаждение и нагревание исследуемых образцов производят по возможности с одинаковой скоростью и при непрерывном перемешивании. Перемешивание расплава обеспечивает образование мелких кристаллов, что способствует сравнительно быстрому приближению системы к состоянию равновесия и выравниванию температуры во всем объеме исследуемого вещества. При проведении исследований этим методом не требуется сложной аппаратуры. Исследования до 500º С можно проводить в пробирке из тугоплавкого стекла, которая помещается в другую пробирку большего диаметра, на последнюю в качестве нагревателя намотана спираль из нихромовой проволоки . Во внутреннюю пробирку помещают исследуемое вещество или смесь веществ, измеритель температуры (термопару, термометр) и мешалку. Нагреватель, намотанный на внешнюю пробирку, служащую, таким образом, печью, подключают к электрической сети через лабораторный автотрансформатор и амперметр для контроля величины тока, проходящего через нагреватель.
Рис .6 Схема установки для визуально-политермического исследования диаграмм состояния. Для более высокоплавких систем (выше 500–600º С) применяются тигли, изготовленные из материала, инертного к исследуемым веществам. Измерение температуры производится при помощи термопары с милливольтметром. Расплав перемешивается с помощью мешалки, инертной к исследуемому веществу, например, платиновой, и сверху освещается ярким пучком света для того, чтобы легче было наблюдать за образованием и исчезновением кристаллов. Печь закрывается для предотвращения конвекционных токов, освещение и наблюдение производится при помощи оптических призм. Охлаждение расплава при этом методе достигается путем отключения источника нагрева.
Рис.7 Схема установки для визуально-политермического высокотемпературного анализа. Получив таким методом ряд значений температур при соответствующих изменениях концентрации, строят диаграмму плавкости, откладывая на одной из осей координат концентрацию, на другой – температуру. Визуально-политермический метод применим при изучении диаграмм состояния систем лишь в том случае, когда расплав прозрачен, что имеет место для многих органических веществ и неорганических солей. Метод неприменим для определения температур конца кристаллизации и превращений вещества в твердом состоянии. Недостатком метода является и то, что возможны ошибки, вносимые экспериментатором при визуальном наблюдении (субъективные ошибки), а также ошибки, возникающие из-за большого перепада температур в самом исследуемом веществе. Несмотря на указанные недостатки, этот метод в виду его простоты, широко применяется для быстрого определения температур начала кристаллизации и построения линии ликвидуса. Метод построения кривых время – температура. Сущность метода заключается в построении кривых время – температура по данным изменения температуры в процессе равномерного нагревания или охлаждения исследуемого вещества или смеси веществ через равные промежутки времени. Для этого исследуемое вещество (смесь веществ) вносится в соответствующий сосуд, помещенный в нагревательное устройство (например, в печь сопротивления); в сосуд с исследуемым веществом вводится измеритель температуры: термопара, термистор, подключенные к измерительной системе. Затем содержимое тигля нагревают выше температуры плавления, после чего нагрев выключают и через равные промежутки времени записывают температуру до полного затвердевания расплава. Если ожидаются фазовые превращения вещества в твердом состоянии, то его охлаждение производится до температуры, которая несколько ниже температуры ожидаемого превращения. Затем производится равномерный нагрев исследуемого вещества и также через равные промежутки времени записывается температура. На основании полученных результатов строят кривые нагревания (охлаждения), откладывая по оси абсцисс время, по оси ординат – температуру. Определяют соответствующую температуру для интересующих участков кривой, отвечающих фазовым превращениям в исследуемом веществе. На полученных кривых им соответствуют изломы, изгибы, площадки, которые в случае значительных величин теплоты превращения будут выражены четко. Если величины теплоты превращений исследуемых веществ незначительны, то определение температур фазовых превращений по изломам на кривых время – температура, построенных по данным визуального отсчета температур, может оказаться затруднительным, а иногда и невозможным. Для увеличения разрешающей возможности метода в этих случаях применяются высокочувствительные термопары, а для отсчета температуры – зеркальные гальванометры. Также можно увеличить чувствительность метода за счет правильного подбора скорости нагрева (охлаждения). Известно, что чем меньше скорость нагрева (охлаждения), тем более четко будут выражены отрезки кривых, отвечающих фазовым превращениям. Однако существуют оптимальные условия нагрева (охлаждения), при которых особенности кривых выражены наиболее отчетливо. Оптимальная скорость подбирается эмпирически, если она заранее неизвестна из аналогичных опытов. Эта величина зависит от теплоемкости, теплопроводности и массы исследуемого вещества. Из сказанного следует, что определение температур фазовых переходов при этом методе не всегда может быть надежным, так как нередко величины тепловых эффектов оказываются слишком малыми, чтобы вызвать более или менее резкое изменение хода кривой температуры. Даже в тех случаях, когда эти эффекты достаточно велики, визуальное определение температуры часто приводит к ошибкам вследствие субъективного характера отсчета температуры и времени. К тому же, этот метод позволяет определить с достаточной точностью лишь температуры начала и конца кристаллизации, определение же температур превращений в твердом состоянии часто практически невозможно в виду малых тепловых эффектов, сопровождающих эти превращения. Дифференциальный термический анализ Дифференциальный термический анализ (ДТА) считается одним из чувствительных и совершенных методов термического анализа. В основном чувствительность этого метода обусловлена большой разрешающей способностью приборов, регистрирующих изменение температуры и непрерывностью этих измерений. Этот метод основан на автоматической регистрации температуры и разности температур между исследуемым веществом и эталоном, то есть веществом, не имеющим фазовых превращений в изучаемом интервале температур, а также с теплопроводностью, близкой к теплопроводности исследуемого вещества. В настоящее время он почти вытеснил метод с визуальным отсчетом температуры. Если в исследуемом веществе фазовые превращения не имеют места, то при его нагревании кривая простой записи, то есть кривая, дающая зависимость время – температура, должна равномерно подниматься вверх, удаляясь от оси времени, а кривая дифференциальной записи (кривая, дающая зависимость время – разность температур) двигаться параллельно оси времени. Первая кривая укажет на равномерно повышающуюся температуру в образце, а вторая – на разность температур образца и эталона. Эта разность при отсутствии превращений должна быть равна нулю. При наличии превращения, сопровождающегося поглощением или выделением теплоты, простая и дифференциальная запись зафиксируют изменение хода кривых. На первой кривой (простая запись) это выразится заметным изломом (часто слабым), а на второй (дифференциальная запись) – резким отклонением. В случае поглощения теплоты системой (эндоэффект) кривая отклоняется вниз, в случае выделения (экзоэффект) кривая отклоняется вверх. После превращения разность температур вещества и эталона снова должна стать равной нулю (в идеальном случае) и дифференциальная запись вновь должна идти параллельно оси времени. На рис. 17 представлены кривые, записанные на саморегистрирующем приборе (пирометре) в координатах время – температура (τ – t) и время – разность температур (τ – ∆t). Как видно из рисунка, кривая температуры в начале нагревания несколько отклоняется от прямой линии, соответствующей температуре печи. Начиная с τ1, разность между температурой печи и температурой исследуемого вещества становится величиной постоянной, температурный градиент ∆t' не зависит от времени. Другой характеристикой данной кривой является величина ∆τ, которая показывает время, необходимое для прохождения теплоты от печи к образцу. Кривая разности температур (дифференциальная запись) в начале нагрева также несколько отклоняется от своей нулевой линии. Начиная с τ1 линия разности температур становится параллельной оси времени, ∆t – постоянна.
Рис.8 Термические кривые: ODВ – кривая температуры, OА – линия, соответствующая нагреву печи. СD'F – кривая разности температур, СЕ – нулевая линия дифференциальной записи. К – соответствуеттемпературе начала процесса (К' на дифференциальной кривой). М (M') – температуре конца процесса.
|