| ПОЗНАВАТЕЛЬНОЕ Сила воли ведет к действию, а позитивные действия формируют позитивное отношение Как определить диапазон голоса - ваш вокал Игровые автоматы с быстрым выводом Как самому избавиться от обидчивости Противоречивые взгляды на качества, присущие мужчинам Вкуснейший "Салат из свеклы с чесноком" Натюрморт и его изобразительные возможности Применение, как принимать мумие? Мумие для волос, лица, при переломах, при кровотечении и т.д. Как научиться брать на себя ответственность Зачем нужны границы в отношениях с детьми? Световозвращающие элементы на детской одежде Как победить свой возраст? Восемь уникальных способов, которые помогут достичь долголетия Классификация ожирения по ИМТ (ВОЗ) Глава 3. Завет мужчины с женщиной
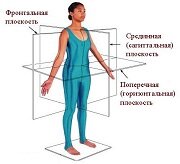
Оси и плоскости тела человека - Тело человека состоит из определенных топографических частей и участков, в которых расположены органы, мышцы, сосуды, нервы и т.д.
| Іштен болатын коагулопатиялар
Іштен болатын коагулопатиялардың 94-96%-н А, В гемофилиясы мен Виллебранд ауруы қүрайды. Қалғандары (С-гемофилиясы — ХІ-фактордың, Хагеман кеселі — ХП-фактордың, парагемофилия — Ү-фактордың, диспротромбинемиялар — II, ҮП, X-факторлардың, ал афибриногенемия, дисфибриногенемиялар, афибриназемия — ХШ-фактордың кемістігінен) сирек кездеседі; олар салыстырмалы диагноз жүргізу үшін керек. Гемофилия Гемофшіия (haima — цан, phiiia— бейімділік) —рецессивті, Х-хромосомамен циылыса берілетін іштен біткен ауру; кеселдің айқын сипатына ҮШ-не IX-плазмалык, коагуляциялық факторлардың кемуінен қанаққыштық жатады. Көбіне ер балалар ауырады. Дүниежүзілік денсаулық қорғау үйымының сарапшылар комитетінің мәліметі оның мөлшері калыпты деңгейдің 1-2%-на ғана тең болса, орташа түрінде — 2-5%, Буыңдарға қан құйылу — гемартроз— аурудың ең көрнекті сипаты, ол мүгедектікке әкеледі. Көбінесе ірі буындар жаракаттанады: тізе, тірсек, білек т.б. Бұл кұбылыста 3 кезен байқалады: гемартроз, артрит, анкилоз. Гемартроз көбіне болмашы закымнан сон. басталады: күшті ауырсыну, буынның ісінуі жэне көлемі ұлғайып, флексия түрінде қалуы. Асқынбаған кезде буынның калпына келуі 1-3 аптаға созылады. Әдетте, гемартроз қайталағанда осы буын(дар) қайта қатысады: бұл орайда ревматоидтык артритке ұқсас аутоиммундық әсер байқалып, сүйекте де өзгеріс болуы мүмкін. Гемофилия кеселінде қан ағысы зақымнан кейін іле-шала емес (тіс жұлған соң), 1-4 сағат өткен соң басталады; қан жуық арада токтамайды, шамалы емге бой бермейді. Мысалы, саусақтан жалпы қан сынағын алудағы зақым, тері, инемен етке дәрі жіберудің әзі сағат, тәулік бойы тоқтамайтын айқьш кан ағысына жалғасуы мүмкін. Гемофилияға мұрын, ауыз, сиректеу ішек-карын шырыш қабаттарынан, бүйректен болатын үзақ кан ағысы тән. Кеңірдек шырытынан аққан қан өте катерлі, себебі тыныс жолының жедел кептелуі трахеотомия кажеттігіне әкеледі. Осы сияқты мойын төңірегіндегі закым, ми мен оның кабығына қүйьшған қанның орталық нерв жүйесіне (ОНЖ) келтіретін әсері кейде өлімге әкелуі мүмкін. Егер VШ-фактор мөлшері қалыптан 15% кем болса, кеселдің анық бейнелері білінбеуі де мүмкін (латентті түрі): бұл тіс жұлу, зақымдану, операциядан соң ұзақ қан ағысқа бейімділік түрінде ғана байқалады. Қанның YIII-факторы деңгейіне байланысты геморрагия ауырлығы (ДЦҚҮ сарапшылары Комитеті мәліметі)
Жас асқан сайын геморрагиялық синдром ауырлығы жеңілдейді. Криопреципитат не антигемофилдік глобулинмен жиі емдегенде қанда осыларға қарсыденелер түзілуіне байланысты (ҮІП-факторға) аурудың 5-10%-да кесел ингибиторлытүрге ауысады: әрине, бүл жағдай кеселдің енді ауыр ағыммен жүретінін көрсетеді. Қанақкыштықтың негізі — қан ұюдың 1-ші мерзіміндегі бұзылысқа қарсы (ҮІІІ, X, XI) факторлардыц кемістігінен—тромб түзілмеуіне байланысты. Әдетте, гемофилияға қанұю мерзімінің ұзаруы (10-30 минутқа дейін) тән, кейде бұл бірнеше сағатқа созылуы мүмкін. Диагнозы мен ажырату диагнозы.Диагноз қоюда шежіре тарқату (шеше жағындағы ер адамдардағы қанаққыштық), анамнез жинау, Ли-Уайт бойынша қанұю уакытының қалыптан тыс (8 минуттен ұзак) ұзаруы, қан ұюдың 1-мерзіміндегі өзгерістері (протромбиннің керек мөлшерінің азаюы — қалыпта 80-100%), ҮІІІ не ІХ-фактор деңгейінің ете төмен көрсеткіштері көмектеседі. Бұдан басқа көрнектілері: 1) рекальцификация уақытының азаюы; 2) тромбопластин түзілісінің бүзылуы. Аурудың анық көрінісі диагноз қоюда қиындық келтірмейді. Зақымнан соңғы ұзак қан ағу, жайылмалы гематома мен гемартроздың ер балада көрінуі — кеселдің бұлтартпас сипаттары. Гемостаздың тромбоцитарлық буынының бұзылыстары ретіндегі қанаққыштық түрі өзгеше: терідегі геморрагиялық синдром петехий, кішігірім канталау (экхимоз) түрінде көрінеді (гематомалар сирек), бұларға гемартроз тэн емес, негізгі симптомы - қайталамалы мұрын қанауы (гемофилияда ете сирек). Аурудың бұл тобында кішігірім операция (тіс жүлу, аденотомия т.б.) іле-шала қан ағуға ұласады (гемофилияда канағу басталу уакыты көп кейін). Шежіре тарқатуда қанаққыштықгың доминантты түрде берілуі анықталады. Эндотелий бүтіндігіне арналған сынақтар оң болады («қысу», «шымшу» симптомдары, бұлар гемофилияда кездеспейді), кан ағу уакыты да ұзарады (бұл да гемофилияға жатады). Жүре пайда болған вазопатияларда коагуляция сынақтары, әдетте, өзгермейді (жалпылама кантамырлық ұю болмаса); отбасылық қанакқыштық белгі кездеспейді. Гемофилияның орташа көрінісінде Вилебранд кеселін естен шығармаған жөн (қанаккыштық доминантты түрде беріледі, қанағу уақыты өте ұзақ; тромбоцитгердің шынайы адгезиясы, ристоцетинмен агрегациясы өте төмен). Гемофилияның С түрі (Розенталь ауруы, ХІ-фактор кемістігі)жеңіл түрде өтеді, сирек кездеседі, өздігінен қанау сирек (мұрын); гемартроз, миға, ішкі ағзаларға қан құйылу болмайды. Ауру аутосомды-доминантты түрде беріледі, әрі екі жынысқа бірдей. Диагноз қоюда гемофилияға тән қанұюдың 1-ші мерзіміндегі өзгерістерге қоса, ХІ-фактордың кемістігі табылуы көмектеседі. Парагемофилия(Оврен ауруы, Ү-фактор кемістігі) аутосомды-доминантты типпен беріліп, тек гомозиготты «тасушыларда» кездеседі. Негізгі сипаты — қайталамалы мұрын қанауы, аз ғана зақымнан соң гематома, қанағу, қыздарда меноррагия (өте күшті болуы мүмкін). Гемартроздар сирек. Диагноз қанұюдың I және П-мерзіміңдегі өзгерістер көрінісі мен Ү-фактордың (проакцелерин) кем деңгейін анықтау арқьшы қойылады. Емі А гемофилиясына ұқсас. Стюарт-Проэур ауруы(Х-фактор кемістігі) аутосомды-рецессивті жолмен беріледі; фактор өте төмен деңгейінде ауру нәресте кезден-ақ білінеді (кіндіктен, зақымданған, кесілген жерден кан ағу), Гемартроз сирек. Диагноз қанұюдың І-ІІ-мерзіміндегі өзгерістер мен Х-фактордың кем деңгейін анықтау арқылы қойылады. Протромбин мен проконвертин(II жэне ҮП-факторлар) жетімсіздігі аутосомды-рецессивті жолмен беріледі; көрінісі гемофилияның орташа ауыр дәрежесіне сай, бірақ өздігінен шырыш қабаттардан (мұрын, жатыр, бүйрек, ішек-мұрын) қан кету белгісі айқын. Зақымдалғаннан кейін буын, ішкі ағзаға қан құйылу, гематомалар болуы мүмкін. Диагноз қанұюдың П-мерзіміндегі өзгерістері мен II-, ҮІІ-факгорлардың кем деңгейін анықтау арқылы қойылады. Емі - плазма құю, РР8В (II, ҮІІ, IX және Х-факторлар концентраты) дәрісі 2-3 күн. Афибриногенемия. Кешенді миокалъцин(«Сандоз» фирмасы) — қанда калъцийтонин төмен кезде: лазер (курсы 3-10 сеанс); кей науқастарға операция — субтотальды синовэктомия. Елизаров аппаратын қоддану жатады. Тіс жұлу керек жағдайда жарты сағат бұрын криопреципитат (15-20 Б/кг есебінде), сонан соң 6 сағаттан кейін осы құюды қайталайды; кейін 3 күн бойы күн сайын. содан соң күнара тіс орны жабылғанша. Аминокапрон қышкылы апта бойы. Баска операцияда да осы негізде, тек алғашкы 2 құю мөлшері көрсетілгендегіден 2 есе артық (50 Б/кг есебінен). Ингибиторлық гемофилияны емдеуде қан тоқтату үшін криопреципитат қалыты мөлшерден 1,5-2 есе артық болуы керек. Ауыр түрінде иммундық тромбоцитопения, лейкопения, эозинофилия болуы мүмкін. З.С.Баркаған бұларды антифосфолипидтік синдромдамуымен (плазма фосфолипидтеріне қарсыценелердің аса мол түзілуі) түсіндіреді. З.С. Баркаған ингибиторлық гемофилияда (1996) емнің 3 вариантын ұсынады: 1) 3 күн бойы рекомбинантты ҮІП-фактор 100-300 бірлік/кг тэулігіне (80-150 бір/кг 2 сағат бойы, сосын 4-7 бір/кг/сағатына преднизолонмен қоса (1мг/кг тәулігіне). Өмірге кауіпті қан кетуде: 2) протромбин комплексі факторларының концентраты (РР8В); 3) белсендірілген ҮП-фактор енгізу. Криопреципитаттың көп мөлшеріингибиторлар түзілісін тежеуі мүмкін. Кей орталыктарда плазмаферез,десмопрессин қолданылады. В гемофилия еміне антигемофилдік плазмадан басқа лиофилді РР8В концентраты (Франшя), ППСБ (Ресей)-П, ҮІІ, IX, Х-факторлар кешені (ем бойында ЖҚҰ с-мы болатынын ескеру жөн), бебулин Тим-4 (ІХ-фактор, бумен өнделген). Қазір гемофилияның бұл түрінде ІХ-фактордың коммерциялық концентраты колданылады (Соnуnе, Рrорlех — АҚШ). Соңғы жиналған мәліметтер бойынша шет елдерде (АКШ) ҮІП-фактор мөлшерін көтеретін дәрілер қолданылуда (гемофилиянын жеңіл және орташа ағымында — десмопрессин, даназол, новосевен). Диспансерлік бақылау. Учаскелік педиатр гематологпен бірігіп жүргізеді. Ауруға қарсы егу және мектепте физкультура (дене тәрбиесі) сабағынан босатылуы тиіс. Дегенмен де бұлшықетті қатайту үшін баланың дене шынықтырумен айналысқаны жөн. Суық тиген кезде ацетилсалицил қышқылы, индометацин, вольтарен, ибобруфен тектес дәрілерді қолдануға қатаң тиым салынады. Қызуды түсіруге парацетамол беруге болады. Банка қоюға да болмайды - өкпеде геморрагия болуы мүмкін. Бала тәрбиесінде көбіне зақымданудан сақтану жағын ойластырып, өскенде қолайлы мамандық алу жайына икемдеу жөн. Алдын алу жолы. Ауру жазылмайды. Қанағудың алдын алған іс. Болжамы. Қазіргі ойластырылған ем жолдары сәттілігіне жөн, бұл үшін ауру ай сайын криопреципитат қабылдауы тиіс байланысты өмір сүру мүмкіндігі мейлінше жоғарылады. (1975) бойынша 10000 ер жынысты нәрестелер ішінде 0,5-1 А-гемофилиялы ауру табылса, В-гемофилия одан сиректеу (0,5) кездеседі. Патогенезі.Науқастардың 80%-да Х-хромосомадағы генге байланысты, ол плазмадағы ҮПІ-фактордың белсенділігін қамтамасыз етеді (АГФ, гемофилияға карсы фактор). АГФ молекуласы, мүмкін, 2 жақты болуы; жоғары молекулалы (ҮІП: КАг), құрамында Виллебранд факторы және антигендік детерминанты бар; екіншісі, аз молекулалы (ҮІІІ: ВФ), прокоагулянттық немесе қан ұйытарлық әсері бар. Сау адамдарда бұл екеуінің плазмадағы ара-қатынасы тұрақты. Гемофилияның А түріндегі науқастың қанындағы ҮІИ : КАг, ҮІІІ : ВФ мөлшері қалыпта болса, ҮІІІ : К өте төмен (0-5%-ға дейін). Ауыратындар – көбіне ер жынысты балалар, себебі олардың аналары — ауру, дертті геннің «тасушылары» (кондуктор). «Тасушыларда» ҮІІІ: КАг, ҮІІІ: ВФ мөлшері қалыпта, ал ҮІІІ: К мөлшері 50-60%-ға төмен. Бұл жағдай қазіргі кезде «тасушы» - әйелдерді дәл анықтап, генетикалық анықтама беруге мүмкіндік туғызады (шаранада А гемофилияны 8 апталықтан кейін). Коагуляцияға катысы бар екі белоктың кодын қарастыратын ген (ҮІІІ : К, ҮІІІ: КАг) X хромосомада (Хg28)орналасқан, ал ҮІП : ВФ түзілісін аныктайтын ген — 12-хромосомада. ҮІІІ : К ген 1989 ж. бөлінді, 186 мың негізден тұрады. Шамамен гемофилияның 25% - спонтандыкмутация сандары. Мұныңжиілігі: А-1,3*10~5, В-6*10~7. Гемофилияның А түрінің 25-30%-да, В түрінің 10%-да — отбасылық анамнез болмайды (яғни кан ағуға бейімділік, қанақкыштык), бұны геннің жаңа мутациясымен байланыстырады (спонтандык түрі), кейін бұл да классикалық А сияқты тұқым қуалайды әрі ауыр ағыммен жүреді. Гемофилияның С түрінде тұқым қуалау аутосомды-рецессивтік жолмен жүреді; А жэне В түрінен өзгешелігі — жынысқа қарамайды; әрі геморрагияға бейімділік келесі буынға ер мен әйел аркылы бірдей тарайды. Әйелдер гемофилиясы өте сирек кездеседі: 1) әкесі гемофилиямен ауру, ал шешесі — кондуктор; 2) тестикулярлық феминизация аурулары; 3) гемофилия кондукторында Тернер синдромы болуы. УІП-фактор түзуге қатысты X хромосомадағы ген Г-6-ФДГ ферментін түзетін жэне түс ажыратуға байланысты гендермен қатар орналасқан. Осы себепті гемофилиямен ауыратындар мен оны тасымалдаушы әйелдерде түсті ажырата алмау (дальтонизм) мен Г-6-ФДГферментінің кемістігі жиі кездеседі (гемофилия кондукторы - әйелдердін 90%-н осыжолмен анықтауға болады). Клиникасы.Гемофилия сипаты кез келген жаста көрінуі мүмкін. Ең ерте белгілері байланған кіндіктің қанауы, кефалогематома, теріасты канталауы т.б. түрінде болуы мүмкін (ҮІП-фактор плацентадан өтпейді). Алғашкы белгісі тіс шығарда, көбінесе бала жүре бастаған кезде көрінеді. Қанталау көбіне комакты, жайылуға икем болады. Қанталаудың жиі түрі — гематома (ет арасына қан кұйылу) көбіне теренде болып, ауырсыну айкын білінеді. Төгілген кан кеп уақыт сұйық күйінде калатындықтан ет талшықтары бойымен тез тарайды. Гематома өте жайылмалы болғанда нерв жүйелері мен үлкен қантамырына закым келуі мүмкін (паралич, парез, гангрена, күшті ауырсыну). Кеселдің көрінісі (ауыр, жеңіл) ҮІП-фактордың мөлшеріне тікелей байланысты: ете ауыр түрінде фибриназа кемістігі (ХП-фактор). аутосомды-рецессивті жолмен беріліп, нәресте кезден-ақ кіндіктен ұзақ қанағумен білінуі мүмкін. Кейін геморрагиялық диатез азаяды, бірак өздігінен мүрын қанауы, зақымнан соң қачағу тоқтай қоймауы, түрлі дәрежедегі пурпура байқалады. Афибриногенемия жараның үзақ жазылмауы түрінде білінеді. Афибриногенемия ұзақ уақыт (бірнеше күн) қанның ү_йымауымен, ал афибриназемия қансоқтасының мочевинаның 5% ертіндісімен еруі арқылы анықталады. Емі - эр апта сайын плазма кұю (керек жағдайда). С протеин кемістігі, К витамин тәуелді гликопротеин;мұның белсенді түрі (тамыр қабатыңда тромбин-тромбомодулин кешенімен белсеңціріледі) Ү, ҮІП-факторлардың белсенді формаларын тежеп, фибринолизді үстемелейді; клиникасы нэрестелік кезенде ақ ауыр тромбоз, тері геморрагиясымен білінеді немесе гепаринге көне қоймайтын ЖҚҮ синдромы дамиды. Қажеті антикоагулянттар, анаболикалық стероидтар (даназол), ал жіті кезеңінде — жаңа тоңазытылған плазма. Протеин кемістігі не антитромбин III(аутосомды рецессивті типпен беріледі) тромботикалық аурумен көрініс табады, диагнозы арнайы тексерулерді қажет етеді. Гемофилия Д3 (ХП-Хагеман факторының кемістігі) өте сирек кездеседі. Қазірғі кезде гемо і).филия диагнозын ер бала дүниеге келгенше де қоюға болады Емі. Гемофилия емінің өзгешеліктері бар: 1) дәріні етке жіберуге қатаң тиым салынады (барлык дәрі тек қанға не ішуге беріледі); 2) ішкі ағзаларға кан құйылу күдігі. кандай дәрежедегі болмасын қан ағысы, буын ісінуі мен ауыру сезімі болса, уакьгт создырмай (түн ішінде болсын) антигемофилдік дәрілерді міндетті түрде қолданады; 3) тері зақымданған кезде де тап осылай істеу керек; 4) әр тоқсанда гемофилиямен ауру бала осы ауруды емдеу тәжірибесі бар стоматологқа көрініп тұруы керек; 5) кез келген операция тек антигемофилдік глобулинді алдымен колданудан соң жасалуы тиіс. Ем 2 түрлі бағдарламамен жүргізіледі: 1) үздіксіз трансфузия; 2) симптоматикалық трансфузия. Бірінші бағдарлама гемофилияның ауыр және орта түріне арналған. А гемофилиялы ауруға әрбір 10-14 күн сайын қанға ҮІП-фактор преципитаты құйылады (1-3 жастарға — 200 бірлік, 4-7 жастарға — 400 Б, 7-10, одан жоғары жастағыларға — 600 Б), В гемофилиясында - ІХ-факторы бар таза плазма (1-5 жастарға ІХ-фактордын 150 Б, 6-10 және жоғары жастағыларға — 300 Б). Екінші бағдарлама бойынша кан құйылудан не үлкен зақымдалудан соң іле-шала ҮІІІ, ІХ-факторлардың концентраты колданылады. А гемофилиясында қанға криопреципитат 20 Б/кг есебінде құйылса, В гемофилиясында РР8В (II, ҮП, IX, X факторлар концентраты) 15 Б/кг есебінде немесе осы мөлшерде плазма құйылады. Емді 8 сағаттан соң қайталаған жөн. Және ескертетін жай: көрсетілген дәрілер жылы күйде, жылдам құйылуы тиіс. Қан тоқтағанша немесе гемартроз азайғанша осы ем кемінде 2-4 күнге созылғаны жөн. Қан аккан жерге гемостатикалык сүзгі, тромбин қойып, катты байлап, суық басу керек, зақымданған жерге емшек сүтін жағады. Гемартроз: жедел түрде алғашкы 4 күн бойы криопреципитат не антигемофилді плазма кұяды. Буын 2-3 күнге қалыпты жағдайда әрі қозғалыссыз болғаны жөн. Буынға кұйылған қанды сору (пункция) хирургке жүктеледі. Виллебранд ауруы Виллебранд ауруы (ангиогемофилия)— тұқым қуалайтын жедел, әдетте аутосомды-доминантты типпен беріліп, жоғары қанаққыштық, қанағу уақыты ұзаруымен, сандық және сапалық кемістікті плазмалык, Вилленбранд факторы (ВФ) және тромбоциттердің өте төмен (не болмауы) адгезия (шыны) мен агрегациясы (ристомицинмен) қосарлануымен ерекшеленеді. Жиілігі - 100.000 адамға 10 аурудан. Патогенезі. Ауру жайлы алғашқы мәлімет берген Э.ф.Виллебранд (1926). Бұл кесел кезінде коагуляция кемістігі (ҮПІ-фактор төмен) мен қантамырлык-тромбоцитарлық бұзылыстар (қанағу уақыты ұзаруы) орын алады. Виллебранд факторы түзілісін реттейтін аутосомды ген 12-хромосомада орналасады, оның нуклеотидтерінің орналасу тізбегі 1990 ж. анықталды. ВФ ҮІІІ: К үшін тасымал-белок, тромбоциттердің микрофибриллаларына адгезия үшін кажет. Адгезия стадиясында ол тромбоциттердің 1Ь гликопротеинін жарақат тамырдың субэвдотелиалды құрамдарымен байланыстырып, агрегация кезеңінде ІЬ гликопротеин/ІПа тромбоцит бетінде болады. ВФ негізінде тамыр эндотелиінің (Паллади денешіктері) арнайы гранулаларында түзіледі. Виллебранд ауруының негізгі 3 типі бар: 1) аздаған сандық кемістік (аурулардың 70-80%) 2)ВФ-ның сапалық кемістігі (аурулардың 10-12%,) аурудың ІІА вариантында үлкен және аралық мультамерлер түзілмейді; ПВ — тромбоцитгердің в гликопротеиніне жоғары аффиндік болады; ІІМД — ҮШ : К аффиндік төмен не тромбоциттердін функционалдық белсенділігі төмен. 3) ҮІІІ: ВФ толык саңдық кемістігі (аурулардың 3-5%); Барлық варианттарда түрлі дәрежеде тромбоциттердің ристоцетинмен агрегациялық белсенділігі төмен, бірақ ІІВ типінде олар кейде қалыпты да болуы мүмкін. Клиникалық сипатының әртүрлілігі кей ауруларда коагуляциялық, басқаларда тромбоцитарлық кемістік басымдығына байланысты. Клиникалық көрінісі.Аурудың ауыр ағымы (ҮІІІ-ф қалыптың 5%-нан төмен) гемофилияға өте үқсас. ҮІП-фактор көбірек кезінде басты орынға тамыр-тромбоцитарлык қанаккыштық шығады: ауык-ауық көптеген тері геморрагиялары, мұрын, жатыр, асқазан-ішектен қан кетулер (гематома да мүмкін, мысалы инъекция орны; гемартроздар). Шамалы хирургиялық әрекеттер кезінде (тіс жұлу т.б.) қан кету басталып, бірнеше тәулікке созылып, постгеморрагиялық анемияға әкелуі ықтимал. Жеткіншек жасында кесел ауырлығы төмендейді. Диагнозы.Ауруды анықтау тек бір мезгілде және динамикада ҮІП-фактор деңгейі мен тромбоцитгер қаблетін тексеру кезінде ғана мүмкін, себебі кеселге тән кемістіктер ауық-ауық жоғалуы мүмкін. Айқын кемістіктерге жататыңдары: 1 )қанағу уақытының аса ұзақка созылуы 2)тромбоциттер адгезиясының шыны түтіктері мен шыны тасбихтарға төмендігі 3)қанда ҮІП-фактор аздығы 4)тромбоциттердің ристоцетинмен агрегациясының темендігі Соңғы кемістік өте тұракты. Тромбоциттер саны ылғи қалыпта. Мелена, гематурия, буынға қан құйылу сирек кездеседі. Миға қанқұйылу (2-4%) да, басқа ішкі ағзалармен қатар, өте сирек болады. Көк бауыршамалы үлкейеді (+1-2 см), ол аурулардың 10%-да байкалады (бұл кеселге тән емесканағудан кейінгі амемия не косалқы аурулар болмаған жағдайларда), дене қызуыкалыпта. . Диагнозы мен ажырату диагнозы.Лабораториялык. тексеруде ИТП-ға тән қалыптан тыс өзгерісгер: тромбоцитопения (қалыптағы саны 150-400* 109/л), кан ағу уакытының артуы (Дьюк бойынша, дәлірегі Айви не Борхгревин-Ваалер сынағы), капиллярлар ұстамдылығының кемуі (банка, қыл бұрау ж.б.), сүйек миында болымсыз мегакариоциттердің көбеюі; мегакариоциттерден тромбоциттердің ажырамауы. Ажырату диагнозын жүргізгенде еске алатын жай: мегакариоцитарлық өскіннің тұракты гипоплазиясы, миелоидтық және эритроидтык өскіндер өзгерісінің кан ағуынан кейінгі анемиясыз көрінісі ИТП-ға мүлдем тән емес, мұндай кезде тромбоцитопенияның басқа себептерін іздестіру керек. Қызу көтеріліп, лимфа түйіндерінін өсуі, көк бауырдың аса ұлғаюы, лейкоцитоз, лейкоцитарлық формуладағы көрнекті ығысу, ЭТЖ-ның артуы, диспротеинемия — бұлар да ИТП-ға жат сипаттар, сондықтан бұл жағдайлар жоғарыдағыдай гипо, апластикалық анемия, лейкоз, жүйелі қызыл ноқта, инфекциялық ауруларды, липидоз не ретикуло-эндотелиоздарды еске алуға мәжбүр етеді. ИТП-ны басқа геморрагиялық диатездерден де ажырату керек (Гланцман тромбоастениясы, Виллебранд ауруы, гемофилия, геморрагиялык васкулит). Іштен болатын громбоцитопениялық пурпуралар негізінде тромбоциттердін аз түзілісі не олардың шектен тыс ыдырауы жатуы мүмкін. Гипопластикалық тромбоцитопениялық пурпуралар(амегакариоцитозбен не мегакариоциттер гипоплазиясымен). Әдетте, басқа кемістіктермен кабаттаса жүреді. Аутосомды-рецессивті жолмен беріледі. Болжамы нашар, аурудың жартысынан көбі жасқа толмай қайтыс болады. Бұның негізінде тромбоцитопоэтин түзілісінің кемістігі болуы да мүмкін (доминантгық жолмен беріледі). Іштен болатын тромбоцитолитикалык, пурпуралармикро, макро- жэне нормоцитарлық болады. Бұлардан көбірек зерттелгендері Вискотт-Оддрич синдромы, геморрагкялыктромбоцитодистрофия (Бернар-Сулье синдромы) және Мей-Хегглин аномалиясы. Вискотт-Олдрич синдромынаүштік сипат тән: 1) экзема; 2) геморрагийлер; 3) инфекцияларға зор бейімділік. Бұлардың негізіндегі иммундық кемістік рецессивті жолмен X хромосомымен қиылыса беріледі. Бернар-Сулъе синдромырецессивті-аутосомдық жолмен беріледі, қанаққыштық пурпура ауыр түрде нәрестелік кезден-ақ білінеді. Осы кезде тромбоцитопения анықталып не оның одан әрі төмендеуі болады. Тромбоциттер үлкен, ристоцетин, өгіз фибриногеніне де аггрегация бермейді. Тромбоциттер кемістігінің негізі — орамында (қабатында) гликокальциннінжоқтығы; бұл қабат кұрамында Виллебранд факторының рецепторы болғандықтан олардың адгезивтік, агрегациялык қабілеті төмендейді. Мей-Хегглин синдромындақанаккыштық шамалы, мектеп жасында көрінеді. Тромбоцитопения тромбоциттердің үлкен көлемімен қабат жүреді, нейтрофилдер мен моноциттерде Джоли денесі анықталады. Тромбоцитопеииялардың иммундық жолмен кейбір дәрілерді қолданған кезде сынақтар оң да теріс те болуы мүмкін. Бұл кеселге күдік туған кезде науқасты динамикада тексеру өте мәнді; себебі аурудың І-типінде, еңтексерілген әрі жиі түрінде де, аталған сынақ көрсеткіштері ара-тұра қалыпта болуы мүмкін. Арнайы орталықтарда диагноз қою үшін плазмада ҮІП:К, ҮП:ВФ, ҮІП:ВФаг, ҮІП:ВФ полимері, ристомициннің ең төмен мөлшерімен (0,2-0,6 мг/мл) тромбоциттер агрегациясы; бұларға сау адамдардың тромбоцитерімен А гемофилиялы науқастар ғана жауап береді, ал Виллебранд ауруының I, ІІА, Ш-типтері үнсіз қалады. Көбінесе диагноз қою тек қайтара тексерулерден кейін ғана мүмкін. Виллебраңд ауруының ДНК-диагноздық тәсілі ең сенімді (В.С.Баранов бойынша диагноздық бағасы 100%-ға таяу). Виллебранд ауруы синдромы ДТДА жэне басқа иммундық дерттер кезінде кездесуі мүмкін. Емі.Аурудың І-типінің жеңіл және орташа ауыр түрлерінде (ҮІП:К>5%) тиімдісі табиғи АДГ аналогы — ДДА УР(десмопрессин ацетат-1-деамино, 8-Д-аргинин вазопрессин). ДДА ҮРқанға 0,3 мг/кг изотоникалық №С1 ерітіндісімен (30-50 мл) 10-20 мин бойында қанға енгізіледі. Келесі енгізу 12 сағаттан соң, бірақ 24 сағаттан соң да қайталау қажет (қайта енгізген кезінде сумен улану және гипонатриемия болуы мүмкін). Кеселдің ІІ-В түрінде ДДАҮР енгізу тромбоцитопения беруі мүмкін. Дегенмен гемофилия емінен жеңіл. Виллебранд ауруының тромбоцитарлығынан басқа түрінде жаңа тоңазытылған плазма (15 мл/кг) не криопреципитат (құрамында ҮШ:ВФ бар) өте тиімді. Бұдан соң ҮІП:К факторы белсенділігінің арту мерзімі 2-3 тәулік; ауық-ауык дицинон, кальций пантотенаты мен фитоемін жүргізу. Трансфузия емі кейде меноррагиялар кезінде де қажет. Бала болмауға әсерлі дәрілер пайдалы (инфекундин, местранол ж.б.), мөлшерін төмендетіп, циклдың 21-күніне дейін. Бұдан басқа ПАМБА, Е-АҚҚ 0,22/кг рег оз еттекірінің бірінші күнінен соңына дейін; трансэксамин қышкылы жергілікті және ішке 0,1 г/кг тәулігіне 3 ретке. Гемофилия жэне Виллебранд ауруы кезіндегі қан ағуда тиімдісі криопреципитат жэне АДГ гормоны туындылары мен фибринолиз ингибиторлары (8-АКҚ, ПАМБА). Болжамы. Өмірі үшін оңтайлы. Жүре пайда болған Виллебранд ауруы (Виллебранд синдромы) Виллебранд синдромы ЖҚН, ПОЛИМИОЗРГГ, ЖКҰ с-мы, амилоидоз, диабет; Фелти, Элерс-Данлос синдромдары, көптеген гемотрансфузиялар соңында байқалған (Моntgоmеrу М. еt; аl., 1982, Тоbeleu А., 1984). Бұл синдром даму себебі — ВФ ингибиторы не оның сапалык аномалиясы. Емі. Гемостаз үшін кортикостероидтар, иммунодепрессанттар (азатиоприн), плазмаферез. Тромбоцитопениялар Бұлар біріншік және екіншілік деп бөлінееді. Біріншілік тромбоцитопенияларға жататындары: идиопатиялык тромбоцитопениялық пурпура (Верльгоф ауруы:), іштен болатын (отбасылық), изоиммундық (туа болатыны— бала мен шешесінде тромбоциттер антигендері бойынша келіспеушілік ретінде, трансфузиядан соңғы - қан не тромбоциттер кұйылғанда) трансиммуңды туа болатын (идиопатикалық тромбоцитопениялық пурпура не жүйелі қызыл ноқтамен ауру анасы арқылы болатын нәрестелер тромбоцитопениясы). Екіншілік (тромбоцитопенияларбалаларда біріншіліктен гөрі жиі кодеседі. Олар инфекцнялық аурудың қызған кезінде (көбінесе перинаталдык вирустық инфкцияларда), аллергиялық не басқа аурулардың жедел ағымды гиперреактивті көріністерінде, дәнекер тіндерінің жүйелі аурулары және осы тектес аутоиммундық бұзылыстарда, жалпы қантамырлық ұю синдромдарында, қан түзу жүйесінде, қатерлі ауруларында (лейкоз, гипо-апластикалықанемиялар); іштен болатын тамырлар (гемангиомалар) және заттық алмасу аномалияларында (Гонпе, Ниманн-Пик т.б. аурулары) кездеседі. Іштен болатын тромбоцитопениялық пурпура негізіне жатырдағы нәрестенің тромбоцитарлық антигені жатады (апасында жоқ бұл жағдай популяцияда 2-5%). Бұл анасында сенсибилизация әсері арқылы тромбоциттерге қарсыденелер түзілуіне, олардың бала жолдасына (плацента) еніп, тромбоцитолиз дамуына әкеледі. Бұл жағдай 5000-10000 нәрестенің біреуінде байқалады: алғашкы күннен денеде петехийлср мен кішігірім қызыл дактар пайда болады. Ауыр түрінде (геморрагиялык синдромның көрінісіндс) шырыш қабаттардың канталауы, мелена, мұрын. окж. кіндік қанау ми қанталауы мүмкін. Диагноз қоюда клиника, лабораториялық белгілср мен нәресте тромбоциттернің анасының қан сұйығында агглютинация беру әсері негіз болады. Тромбоцитопения 2-12 аптаға дейін сақталады, бірақ геморрагиялык; синдром көрісін дұрыс емдегенде алғашкы күндері-ак тыюға болады. Іштен болатын трансиммундық тромбоцитопения нәрестелердің (30-50%) анасы идиоиатиялык тромбоцитопатиялық пурпурамен (ИТП) ауырғанда кездеседі. Бұлардың жарғылсында геморрагиялық көрініс болады. Ауру негізі — анасынын тромбоциттерге қарсы денелері мен тромбоциттеріне сенсибилизацияланған лимфоциттерінің жатыр арқылы нәрестеге өтуі. Алғашқы күндері-ак петехийлер, денеде экхимоздар мен қанталау орындары, кейде шырыш қабаттар, мұрын қанауы. мелена байқалады. Көбінесе қанаққыштық сипаты анамнез, клинико-лабораториялық белгілер және анасында тромбоциттергс қарсы аутоденелер мен екеуінде де өз тромбоциттеріне сенсибилизацияланған лимфоциттердің болуы көмектеседі. Айығу 5-12 аптаға созылады. І-Идиопиялык тромобоцитопениялық пурпураға (ИТП) ауысуы аурудың 1-3%-да байқалады Идиопиялык тромобоцитопениялық пурпура (РГТП) — біріншілік геморрагиялык негізі тромобоцитопениялық гемостаздың сан және сапалык жетімсіздігі. Көрнекті белгілері: пурпура (тері асты және шырыш қабаттар қанталауы), шырыш қабаттар қанақкыштығы, тромбоциттер саны аз; мегакариоциттердін сүйек миындағы саны қалыпта не артық; ағымында тромбоцитопениямен аскынуы мүмкін; спленомегалия не басқа жүйелі аурулар байқалмайды. Этиологиясы мен патогенезі. ИТП — отбасылық бейімі бар ауру; науқастарда іштен болатын тромбоцитопатия болады; осы негізде вирустық инфекциялар (ЖРВИ, кызылша, кызамықт.б.), егу, түрлі физикалықне психикалык салмақ, немесе сыртқы жайттар макрофагтардың қорыту қасиетін бү_зып, одан эрі дертті иммундық әсерлерге соктырады - өз тромбоциттеріне сенсибилизацияланған лимфоциттер көбейіп - өсіп, қарсыценелер түзіледі. Қазіргі түсінік бойынша ИТП-ға, әдетте, дертті иммундык өзгерістер тән, сондыктан оны иммунды не иммунды емес деп ажырату кисынсыз. ИТП-дағы тромбоцитопения негізі күдік тудырмайды: көк бауырда тромбоциттер орасан күйреуге үшырайды, бүл жер әрі тромбоциттерге қарсы түзілетін карсыценелер орны. Осылардьвд аркасында тромбоциттер өте аз өмір сүреді (калыптағы 7-10 күн орнына тек бірнеше сағат кана). Тромбоциттер түзілісі ИТП-да жоғары деңгейде, бүны мегатромбоцштер санының өсуі дэлелдейді. ИТП-дағы қанаққыштық гемостаздың тромбоцитарлык буынындағы сан (тромбоцитопения) және сапа (тромбоцитопатия) жетімсіздігіне орай болады. Тамырдың эндотелий кабаты тромбоциттердің ангиотрофикалық эсерінсіз бүзылыска душар болып, осыдан тамырдың өткізгіштігі артып, өзіндік геморрагийлар пайда болады; бүған серотонин (тромбоциттердегі) жетімсіздігі де қосылады. Гемостаздың коагуляция буыныңцағы өзгерістер (тромбопластин түзілісінің кемуі, фибринолиз артуы) тромбоцитарлық буынға қарағанда косалқы деп есептеледі. ИТГІ-да тромбоцитопатия аурудың кез келген кезеңіне тэн, бүл жағдай спленэктомиядан да, тромбоциттер саны қалыпка келгеннен кейін де байқалады. Ұқсас кұбылыс Минковский-Шоффар анемиясы бойында да байқалады: спленэктомиядан кейін анемия жоғалса да микросфероцитоз қалады. Қанаккыштыкгың тағы бір негізі — ретракция бүзылысы, яғни берік тромб түзілмеуі деп есептеледі. Жіктелуі.Ағымы бойынша жедел (6 айдан кем) жэне созылмалы түрге бөлініп, сонғысын кайталауларына карай варианттарға айырады: а) сирек; б) жиі) в) үздіксіз. Кезеңіне байланысты асқыну (криз), клиникалық ремиссия (тромбоцитопения болса да канаккыштық белгісі жок) жэне клиника-гематологиялық ремиссия деп ажыратылады. Клиникалық көрінісіне сай «таза» (геморрагиялық синдром тек теріде ғана) жэне «ылғалды» пурпура (пурпурамен қоса қанағу сипаттары да бар) деп бөлінеді. Клиникалық көрінісі. ИТПкөп жағдайда мектеп жасындағы қыз балалар арасында 2 есеге жуык жиі байқалады. Көбінесе вирустық инфекциядан 2-4 аптадан соң тері, шырыш қабаттары қанталап, қанағу көрінеді. Пурпураның көрнекті сипаттары: 1) полихромдық (бір мезгілдегі терінің түрлі түсі — кызыл-көктсн жасыл не сарғышқа дейін), 2) полиморфтық (петехийден келемді экхимозға дейін), 3) симметрия сакталмауы, 4) бірден, өзінен-өзі (спонтанды) пайда болуы, көбіне түзеді. • ИТП-ның айқын белгісі - қанағу (мұрын, тіс айналасы, пубертаттык жаста пайда болатын түрлері де бар (хинидин, амидопирин, сульфаниламидтер, стрептомицин, карбенициллин, барбитуратгар, кризанол т.б.) Казабах-Мерршп синдромындатромбоцитопения кавернозды гемангиомамен Емі. Тромбоцитопениямен негізіне тығыз байланысты. Изо- және трансиммунды пурпуралы нәрестелер алғашқы 2 аптада донордын емшек сүтімен қоректенуі керек, сонан соң ана сүтіне тромбоциттер санын бақылау бойында қосады. Басқа тромбоцитопенияларда қоректену қалыпты түрде баланың жасына сай жүргізіледі. Режим, әдетте, тек геморрагиялык криз кезінде ғана шектеледі. Кез келген тромбоцитопенияда (тек ЖҚҰ болмаған жағдайда) эпсилон-аминокапрон қышкылы (ЭАКҚ) берілгені жөн (0,05-0,1 г/кг есебінен күніне 4 рет), болмаса тромбоциттердің адгезия-агрегациялық қабілетін арттыратын дәрілер пайдалы (адроксон, этамзилат немесе дицинон; кальций пантотенаты; етке АТФ, оған қоса ішуге магний дәрілері; фитоем). Геморрагиялық криз кезінде ЭАКҚ күніне 1-2 рет қанға не тамшы түрінде берілуі тиіс. ИТП-да глюкокортикоиттар мына жағдайларда тағайындалады: терідегі жалпылама геморрагиялык синдром, шырышты қабаттардан қан ағуы көз қарашығына қан кұйылумен үштасу, ішкі ағзаларға қан құйылу, «ылғалды» пурпураның қан ағудан кейінгі анемиямен аскынуы. Преднизолон 2 мг/кг есебінен 3-4 апта беріліп, бірте-бірте мөлшерін азайтумен тоқтатады. Ұзақ уақыт, айлап беру тиімсіз және көптеген асқынуларға соқтырады. ИТП ағымы 6 айдан аса созылған «ылғалды» пурпура глюкокортикоидтердің қайталама курстарын жүргізуге мәжбүр етсе; жедел ағымды пурпура күдігі туса, -спленэктомия жасалады. Әдебиеттің жинақты мәліметі бойынша ИТП спленэктомиядан соң 85%-ға жуық клиника-лабораториялық ремиссия береді не қанауы мейлінше азаяды. Спленэктомиядан кейінгі кайталау көбінесе көк бауырдың қосалқы бөлігінің операция кезінде қалып қоюынан деп есептеледі, мұны эритроциттерде Хауел-Джоли денелерінің жоқтығы дәлелдейді (соңғылар спленэктомиядан кейін міндетті түрде табылуы тиіс). Спленэктомиядан көмек болмаған жағдайда аз уақытқа глюкокортикоидтар беріліп, ол да пайдасыз болса, амалсыз аптасына 1 рет қанға винкристин жіберіледі (1,5 мг/ м2), эсері 2-4 аптадан кейін байкалады. Спленэктомияны (әсіресе бір жаска дейін) 5 жасқа толғанша жасау қатерлі, сепсис (пневмококктік) болу қаупі 1-2%; сондықтан операциядан кейін жыл бойына айына 1 рет бициллин-5 дәрісін қолдану керек. Соңғы кезде ИТП-ны қанға тамшымен G-Іg-ін (0,4 мл/кг есебінде 2-3 аптаға 1 рет) құю тәсілімен емдеу меңгерілген. Антирезустык, иммуноглобулин (анти-Д-ІgG) Rһ(+) ауруларға 25-75 мкг/кг есебінде 2-5 күн бойы қанға енгізу) созылмалы ИТП-мен науқастардың жартысына көмек береді — тромбоциттер саны жоғарылайды. Спленэктомия жасалғандарға тиімсіз. Интерферон-а - генноинженерлік дәрі (реаферон, интрон-А) 1988 ж. қолданылуда. Тиімділігі 72,5% (15%-толық, немесе тромбоциттердің саны қалыпқа келеді — Е.К.Донюш (1997). 5 жасқа дейін мөлшері 500 000 ХБ/тәулігіне; 5-12 жас — 1 млн ХБ, 12-ден аса — 2 млн ХБ/тэулігіне етке не тері астына аптасына 3 рет 3 ай бойы, әсері 2 аптадан соң-ақ байқалады. Тромбомасса кұю — тиімсіз, бұны тек өмірге қауіпті қан құйылу (ми) кезінде ғана қолданады. Цитостатик өте сирек, тек спленэктомиядан көмек болмағанда ғана, беріледі (винкристин қанға саулатып 1,5-2 мг/м мөлшерде аптасына 1 рет) кей авторлар бұл жағдайда (О.Наііу, М.\УепЫаі, 1988) синтетикалық андроген — даназолұсынады (20 мг/кг, 2 ретке), 1-12 ай бойы. Плазмафарездіңаса тиімділігі жайлы мәлімет бар (Л.Г.Ковалева ж.б., 1989). Алдын алу жолдары. Тромбоцитопениялық пурпураның біріншілік алдын алу жолдары әлі игерілмсген. Қосалқы бағыттары аурудың қайталауы болдырмаумен шектеледі. Егу мәселесі өте сақтыкты кажет етіп, әрі балада өзіндік байыппен жүргізілуі тиіс. Оқушылар дене шынықтыру сабағынан босатылады, күнге күюге болмайды. Тірі вирустық вакциналармен егуге тиым салынады. Геморрагиялык синдромның алдын алу бойында науқастарға тромбоциттердің агрегациялық қабілетіне зиянды дәрілер берілмеуі тиіс (индометацин, кофеин, нитрофурандар, барбитураттар, аспирин, карбенициллин). Болжамы.Қазіргі емдерге байланысты сәтті. Летальдық жағдайлар 2-3%-дан аспайды. Диспансерлік бақылау.Жедел ағымды ИТП-да 5 жыл бойы жүргізіледі, созылмалы түріндс — баланы үлкендер емханасына аударғанша. Егу сенсибилизацияны азайту жолымен аурудың жедел ағымды кезеңіне жыл толғаннан кейін жасалады. Ауруға 3-5 жыл бойына климат ауыстыру тиімсіз. Диета қалыпты. Ауруханадан шығарған соң қан сынағы мен тромбоциттер санын анықтау алғашқы 3 айда 2 апта сайын 1 рет. ал қалған 9 айда әр ай сайын, кейін 2 айда 1 рет жүргізіледі; осы тексеріс әрбір инфекциядан соң да жасалуы тиіс: 3-6 ай бойына қан тоқтату әсері бар фитожинақтарын беру пайдалы (2 апталық курспен, ауыстыру ретімен). Осы емді 2 айлык курспен жылына 2-3 рет қайталауға болады. Жүректік гликозидтерді қолданбайды, себебі олар сол жақ қарыншаның көлемін кішірейтеді. Хирургиялық емде - қарыншааралық перде резекциясы және митральды клапанның протездеуін жасайды. Антикоагулянтты және антиагрегантты терапияны пароксизмальды немесе аритмияның тұрақты формасы бар науқастарға ғана қолданылады. Науқастардың өлімінің негізгі себебі болып, кенеттен аритмияның болуы, қан айналысының жетіспеушілігі, асқынған тромбэмболия. Рестрективті кардиомиопатия (РКМП)- миокардтың этиологиясы нәтижесінде эндокардтың, субэндокардтың және миокардтың (айқын фиброз) морфологиялық өзгерістерін айтамыз. Қарынша немесе 2 қарыншада зақымдалуы мүмкін. Зақымдалған қарынша қуысында облитерация болуы мүмкін. Жүрек жеткіліксіздігінің клиникалық симптомдары болады. Бұл өте сирек кездеседі. 3. Миокардиттер, кардиомиопатиалар. Диагнозы, емі, режим ерекшеліктері, диспансерлік қадағалау. Миокардит дегеніміз- миокардтың инфекциялық, инфекциялы-аллергиялық немесе инфекциялы-токсикалық зақымдануымен болатын ауруды айтамыз. Этиологиясы:Себебі, инфекция болып табылады, көбінесе - вирусты. Миокардит аллергиялық жағдайда, иондық сәулелену, химиялық физикалық әсерлер кезінде және кейбір дәрілік заттарды қолданғанда туу мүмкін. Этиологиясына байланысты миокардиттің түрлері: вирустық (полиомиелит, грипп, қызылша, паратит, герпес, аденовирус вирустары және тағы басқа), бактериялық (дифтерия, скарлатина) жэне тағы басқа. Инфекциялық фактор миокардитке енеді, лизосомалық ферменттердің шығуын зақымдайды (сепсис кезінде). Иммунологиялық механизмде - аутоантиген - антидене реакциясы, иммунды комплекстің қүрылуы, медиаторлардың бөлінуі және қабынудың пайда болуы кіреді. Клиникалық көрінісі:Аурудың ағымына жүру дәрежесіне байланысты: жеңіл, орташа ауыр жэне ауыр болып бөлінеді. Миокардитті тахикардияда, аритмияда және генезі анықталмаған жүрек жеткіліксіздігінде байқауға болады. Миокардит жиІ симптомсыз өтеді. Сирек клиникалық көрініс негізінен инфекциялық процесстен болады. Вирустық миокардитке жиі жоғарғы тыныс алу жолдарындағы инфекциялар, қалтырау жэне асқазан ішек трактісі аурулар кіреді. Шағымдары:жалпы әлсіреу, шаншитын, сыздайтын, қысатын жүрек тұсының ауруы, физикалық күштен кейін ентігуді пайда болуы, жүректің қағуы, жүрек маңайындағы ауытқулар; ауыр ағым сатысында - оң жаққабырға астындағы ауруды сезіну, аяқтың ісінуі, физикалық күште жөтелдің пайда болуы. Объективті: жеңіл ағым сатысында науқастың жалпы жағдайы қанағаттанарлық және ауыр ағымында жағдайы өте ауыр болып келеді. Төсектегі жағдайы мәжбүрлі - жоғарғы қалып (ауыр ағым сатысында). Акроцианоз болуы мүмкін. Пульсі жиіліген (дифтериялық миокардитте брадикардия), қанағаттанарлық немесе толуы әлсіз, аритмиялық (экстрасистолия, мерцательді аритмия). Артериялық қысым төмендеген. Жүрек шекарасы сол жаққа қарай ұлғайған. Жүрек тондары бәсеңдеген немесе естілмейді, аритмиялық (галоп ритмі, экстрасистолия, пароксизмальды тахикардия, мерцательді аритмия), жүрек ұшында систолалық шу (бұлшықет генезінің шуылы), бірінші тон әлсіреген. Ауыр ағымында жүрек жеткіліксіздігі байқалады. Асқынулары: 3 жармалы клапанның жетіспеушілігі, тромбэмболия, сол жэне оң жақ қарыншаның жетіспеушілігі, перикардиттің қосылуы болады. Диагностикасы:ОАК: лейкоцитоз, лейкоцитарлық форманың солға ығысуы, ЭТЖ - ң жоғарлауы. Вирусты миокардитте лейкопения болуы мүмкін. БАК: сиал қышқылының α2 жэне γ - глобулиннің, АсАТ қүрамының жоғарлауы. ИИ: Т лимфоциттердің және Т - супрессордың санының төмендеуін, JgА, JgУ құрамының жоғарлауы, СРП анықталуы. Қанды егуін және басқа биологиялық сұйықтықтың қоздырғышын анықтау. ЭКГ: 8 - Т сегментінің жетіспеушілігінің ауыспалы, спецификалық емес өзгерістері, ритмнің бұзылуы, (ауыр ағымында жүрек блокаторлары), вольтаждың төмендеуі. Кеуде клеткасының рентгенографиясы: жүрек шекарасының ұлғаюы, өкпедегі тұрып қалу өзгерістері. Ауыр жағдайда кардиомегалия. УЗИ: жүрек бүлшық етінің гипертрофиясы анықталынады, қуысының дилатациясы, миокардтың тотальды гипокинезаның белгілері. Миокардтың өмірге қауіпті биопсиясы - қабынудың белгілері. Көптеген жағдайда миокардит жеңіл өтеді және ол жазылумен аяқталады. Ауыр ағымында 50% жағдайы жазылумен аяқталады, ал қалған жағдайда дилатационды кардиомиопатия пайда болады. Жүрек ритмінің бүзылысы кенеттен өлуге алып келеді. Емі: Негізгі - этиотропты терапия жэне асқынуларые емдеу. Әдетте госпитальдау көрсетілген. Бүған төсек режимі, О2 ингаляциясы жэне спецификалық емес қабынуға қарсы препараттарды қолданады. Диета 4-5 г түзды жэне 1 - 1,2 л тэулігіне сұйықтықты шектеу. Этиотропты терапия вирусты гепатитпен шақырылған миокардитте ремантадин (100 мг ішке тәулігіне 2 рет 3 тэулікке дейін); пневмониялық микоплазмамен шақырылған миокардитте - эритромицин (0,5 - 1 г көк тамырға), хламидиямен -доксициклин; әр түрлі бактериямен - антибиотикке сезімталдығымен анықталады - ванкомицин; токсиннің эсері болғанда - антибиотик терапия + антитоксинді жедел енгізу; саңырауқүлақтармен — амфотирицин, фторцитозин; аллергиялық миокардитте антигистаминдік дәрілік заттар,-аллергенді жою үшін; сәулелі миокардитте - кортикостероидтар қолданылады. Науқастарға стероидты емес қабынуға қарсы препараттар (индометацин, ортофен), аминохинолинді препараттар (делагил) қабылдайды. Миокардтағы метаболизмді жақсарту үшін рибоксинді ішке және көк тамырға енгізеді, Е витаминін қабылдайды. Жүрек жеткіліксіздігінің дамуында диуретиктер, жүрек гликозидтерді береді (көрсеткіштер бойынша абайлап беру). Кардиомиопатиялар. Кардиомиопатия (КМП)- миокардтың этиологиясы анықталмаған қабынусыз болатын ауру, бұған кардиомегалия және прогрессирленген жүрек жеткіліксіздігімен сипатталады. Дилатационды КМП (ДКМГІ), гипертрофикалық КМП (ГКМП), рестриктивті КМП (РКМП). Дилатационды ДКМП. Бұл кезде жүректің барлық камерасы ұлғаяды, қабырға гипертрофиясы және миокард функциясының жырылуы төмендейді, нәтижесінде дифференциалдық бұзылыстары және қан айналуының прогрессиялық жеткіліксіздігі дамиды. Этиологиясы. Анықталмаған. Аурудың вирусты табиғаты бар деп ойлайды. Патогенезде Т супрессордың жүре пайда болған деффицитті байқалуы мүмкін, бұл аутоиммунды құбылыстар цитоксикалық, миокардтың иммунокомплекстік бұзылыстың тууына алып келеді. Клиникалық көрінісі: І.Сол жақ жүрекше типі (ентігу, цианоз, ортпноэ, жүрек астмасы және өкпе ісігі), оң жақ жүрекше типі (акроцианоз, бауыр аймағындағы шаншу, оның үлкеюі, асцит, ісіктер, мойын венасының ісінуі) немесе тотальды қан айналуының жеткіліксіздігі айқын көрінген. 2.Кардиомегалия, жүрек тондарының естілмеуі, галоп ритмі, жанама митральды немесе трикуспидальді жетіспеушіліктегі систолалық шуыл. З.Жүректік ритмнің ауыр бұзылыстары (мерцательная аритмия, пароксизмальді тахикардия, экстрасистолия, блокадалар). 4.Тромбоэмбалдық синдром (өкпедегі, көк бауырдағы тромбэмболия). Диагностикалық критериилері: қан айналуының прогрессиялық жетіспеушілігі, кардиомегалия, тромбоэмболдық синдром, босаңсу ритмінің бүзылыстары. Қанның қүрамында креатинофосфокиназаның мөлшері болады. Жас шақта. ИБС белгісінің болмауы, ревматизмнің, жүрек ақауының, қабынулық аурудың, инфекциялық ошақтың б белгілері болмайды. Диагноетиксы: ОАК - ерекшеліксіз. БАК: жоғары кұрамды креатинфосфокиназа, АсАТ. лактатдегидрогеназалар. ЭКГ: сол жақ жүрекшенің үлғаю белгісі. Жүрек рентгеноскопиясы: жүректің барлық бөлімІнің үлғаюы. УЗИ: жүрек қусындағы дилатация, гипертрофияның үстінде дилатацияның болуы. Миокардтың өмірлік биопсиясы: дегенеративті өзгерістер, некроз ошақтары. ДКМП- нің емі: . Емдік режим 2-3 айдан бастап 6 айға дейін төсектік режимді сақтау. Активті режимге көшу үшін миокард дилатацияның толығымен төмендеуі және қан айнлау жетіспеушілігі болмау керек. Шылым шегуге, ішімдік ішуге болмайды. № 10 диета құрамында витаминдер көп болу керек. Жүрек жетіспеушілігінің емі: жүректік гликозидтері бақылап беріледі, себебі ДКМП кезінде миокард гликозидтеріне өте сезімтал болады, Қысқа курс (4-6 күн) бойынша гликозидтік емес, инотропты дэрілік заттар (допмин) беріледі. Перифериялық вазоделитаторлар - ұзақ эсер беретін нитраттар (празозин), АПФ ингибиторы - каптоприл, капозид, энам. Диуретиктер -фуросемид, урегит және триампур, верошпирон препараттарымен қоса. нтиаритмиялық терапия - кордарон, этмозин, этацизин. Антикоагулянттық терапия - іш тері астына гепарин, ацитилсалицил қышқылы (0,125-0,325 г тәулігіне), курантил, трентал. Метаболикалық терапия - поливитаминдер, анаболикалық стероидты емес дәрілік заттар, пиридоксальфосфат, кокарбоксилаза, фосфаден. Хирургиялық ем - жүректің трансплантациясы, яғни ауыстыру. Болжамы - жағымсыз. Өлім себебі: қарыншаның фибриляциясынан кенеттен болатын өлім, қан айналымының тұрып қалған жетіспеушілігі; массивті өкпелік тромбоэмболия. Гипертрофикалық кардиомиопатия (ГКМП)- миокардтың этиологиясы анықталмаған ауру, қарынша аралығындағы жолдар немесе сол жақ қарыншаның қабырғаларында систолалық көлемінің азаюы жэне оның диастолалық функциясының төмендеуі. Клиникалық көрінісі:Жүрек аймағындағы ауруды сезіну шағымдары, естен тану, әлсіздік, бас айналуының ұстамасы, жүрек соғысы, ентігу, төс артындағы ауруларды сезіну. Объективті тексеріс: ұйқы артериясының пульсациясы, тері жамылғыларының бозаруы, жүректің барлық жағынан ұлғаюы (көбінесе солға қарай), жүрек соғысы солға қарай жэне астына қарай ығысуы, жүрек тондары естілмейді, жүректің жоғарғы бөлігінде систолалық шу, кеуденің сол жағында III — ІҮ қабырға арасында, тұрған кезде жиі аритмия болады. Диагностикасы:Анамнезінде: жас кезінде жүректің ауыр түрінде ауруы немесе отбасында кенеттен біреудің өлуі, кардиомегалия, жүректің ұшында және кеуденің сол жағында систолалық шу, қабыну белгілері жоқ, ИБС, ревматизм, жүрек ақаулары, гипертониялық аурудың белгІлері. Лабораторияда - сипатталмайды. Рентгенография, ЭКГ - сол жақ жүрекшенің миокард гипертрофиясының белгілері. УЗИ - қарыншааралық бөгетінің немесе сол жақ қарыншаның қалыңдауы, жүрек камерасының көлемі кішерейеді. ФКГ — жүрек ұшындағы систолалық ромб тәрізді шу естіледі. Емі:Кенеттен өлудің сезінуі. Кенеттен өлудің себебі болып, аритмияның пайда болуы (қарыншаның фибриляциясы, қарыншаның тахикардиясы) жатады. Антиаритмиялық препарат ұғынылады - кордарон (амиодарон), яғни бұл препарат науқастың тірі қалуына жағдай тудырады. Гемодинамиканың жақсаруы. Терапияныңмаңызы зор, қарыншаның созылуының күшеюІне бағытталған және диастола кезінде ол толығымен босаңсиды - адреноблокаторларды қолданады: пропроналол (анаприлин, обзидан) үлкен мөлшерде (120 - 240 - 320 мг тәулігіне) және ұзаққа дейін. Әрбір жеке жағдайға В - адреноблокаторлардың мөлшерін қолдану туралы сұрақтар шешіліп жатыр. Егер анамнезде кенеттен өлу, жақын туысқандарда аритмия болмаса, онда 13 - адреноблокаторларды қолданбауға болады. Са антогонистерді қарынша миокардың және коронарлы артерия перфузиясындағы диастоланың жұмысын жақсарту үшін қолданылады. Диуретиктерді өкпеде венозды қысымның жоғарлағаннан болатын тұнбаны азайту үшін қолданылады. Нитраттарды жэне вазоделитаторларды сол жақ қарыншаның обструкциясының күшейіп кететіндіктен оларды қолданбайды. 4.Балалардағы ревматикалық лихорадка қазіргі заманға сәйкес. Жіктелуі. Клиникасы және ағым варианты. Дифдиагностикасы. Этапы емі. Алдын-алуы. Ревматизм— дәнекер тінінің, оның ішінде әсіресе жүрек, қантамырлар және буындарды зақымдайтын инфекциялық-аллергиялық ауру. Дерттің ерекшедігі—оның қайталап отыруы (рецидивтер) және үдей түсетіндігі. Дер кезінде, тиімді жолмен емделмесе, ауру жүрек қақпақшаларын зақымдайды, миокардта дистрофия және склероз қалдырып, бала жүрегінің жұмысы мен бүкіл қан айналысының жетіспеушілігіне әкеледі. Этиологиясы:Ревматизмнің басталуы (бірінші басталуы болсын, кейінгі атака -рецидив болсын) А-тобының |3—гемолитикалық |